



- La PCR è un metodo utilizzato per amplificare il DNA da una piccola quantità di modello di DNA.La RT-PCR utilizza la trascrizione inversa per produrre un modello di DNA da una fonte di RNA che può quindi essere amplificato.
- PCR e RT-PCR sono tipicamente reazioni endpoint, mentre qPCR e RT-qPCR utilizzano la cinetica della velocità di sintesi del prodotto durante la reazione PCR per quantificare la quantità di modello presente.
- I metodi più recenti, come la PCR digitale, forniscono una quantificazione assoluta del modello di DNA iniziale, mentre metodi come la PCR isotermica riducono la necessità di apparecchiature costose per fornire risultati affidabili.
La reazione a catena della polimerasi (PCR) è una tecnica di biologia molecolare relativamente semplice e ampiamente utilizzata per amplificare e rilevare sequenze di DNA e RNA.Rispetto ai metodi tradizionali di clonazione e amplificazione del DNA, che spesso possono richiedere giorni, la PCR richiede solo poche ore.La PCR è altamente sensibile e richiede un template minimo per il rilevamento e l'amplificazione di sequenze specifiche.I metodi PCR di base si sono ulteriormente evoluti rispetto al semplice rilevamento di DNA e RNA.Di seguito, abbiamo fornito una panoramica dei diversi metodi PCR e dei reagenti che forniamo a Enzo Life Sciences per le vostre esigenze di ricerca.Il nostro obiettivo è aiutare gli scienziati ad accedere rapidamente ai reagenti PCR da utilizzare nel loro prossimo progetto di ricerca!
PCR
Per la PCR standard, tutto ciò che serve è una DNA polimerasi, magnesio, nucleotidi, primer, il modello di DNA da amplificare e un termociclatore.Il meccanismo della PCR è semplice quanto il suo scopo: 1) il DNA a doppio filamento (dsDNA) viene denaturato al calore, 2) i primer si allineano ai singoli filamenti di DNA e 3) i primer vengono estesi dalla DNA polimerasi, risultando in due copie del DNA filamento di DNA originale.Il processo di denaturazione, ricottura e allungamento attraverso una serie di temperature e tempi è noto come un ciclo di amplificazione (Fig. 1).
|
|
| Figura 1.Rappresentazione schematica di un ciclo di amplificazione mediante PCR. |
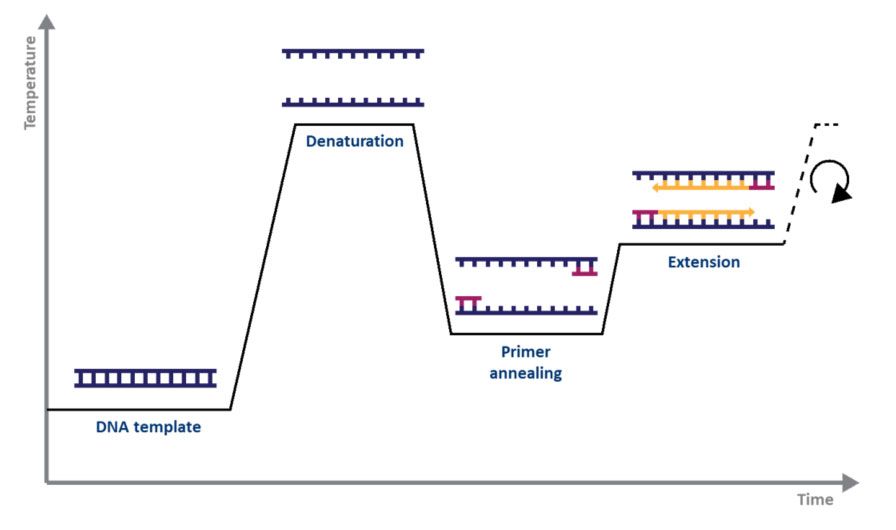
Ogni fase del ciclo deve essere ottimizzata per il modello e il set di primer utilizzati.Questo ciclo viene ripetuto circa 20-40 volte e il prodotto amplificato può quindi essere analizzato, tipicamente mediante gel di agarosio (Fig. 2).
| |
| Figura 2.Amplificazione di un modello di DNA mediante PCR e analisi mediante elettroforesi su gel di agarosio. |
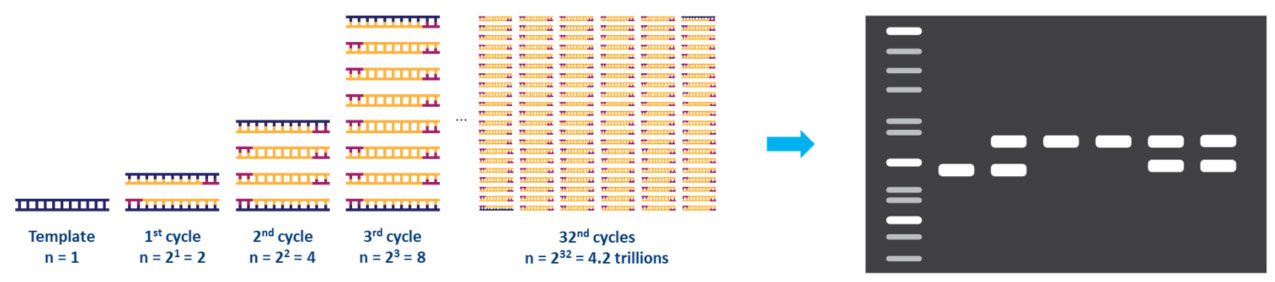
Poiché la PCR è un metodo altamente sensibile e sono necessari volumi molto piccoli per le singole reazioni, si consiglia la preparazione di una master mix per diverse reazioni.La miscela master deve essere ben miscelata e quindi divisa per il numero di reazioni, assicurando che ciascuna reazione contenga la stessa quantità di enzima, dNTP e primer.Molti fornitori, come Enzo Life Sciences, offrono anche miscele PCR che contengono già tutto tranne i primer e il modello di DNA.
Le regioni ricche di guanina/citosina (ricche di GC) rappresentano una sfida nelle tecniche PCR standard.Le sequenze ricche di GC sono più stabili delle sequenze con un contenuto di GC inferiore.Inoltre, le sequenze ricche di GC tendono a formare strutture secondarie, come gli anelli a forcina.Di conseguenza, i doppi filamenti ricchi di GC sono difficili da separare completamente durante la fase di denaturazione.Di conseguenza, la DNA polimerasi non può sintetizzare il nuovo filamento senza ostacoli.Una temperatura di denaturazione più elevata può migliorare questo aspetto e gli aggiustamenti verso una temperatura di ricottura più elevata e un tempo di ricottura più breve possono impedire il legame non specifico dei primer ricchi di GC.Reagenti aggiuntivi possono migliorare l'amplificazione delle sequenze ricche di GC.DMSO, glicerolo e betaina aiutano a distruggere le strutture secondarie causate dalle interazioni GC e quindi facilitano la separazione dei doppi filamenti.
PCR con avvio a caldo
L'amplificazione non specifica è un problema che può verificarsi durante la PCR.La maggior parte delle DNA polimerasi utilizzate per la PCR funzionano meglio a temperature comprese tra 68°C e 72°C.L'enzima può tuttavia essere attivo anche a temperature più basse, sebbene in misura minore.A temperature molto inferiori alla temperatura di ricottura, i primer possono legarsi in modo non specifico e portare ad un'amplificazione non specifica, anche se la reazione viene avviata su ghiaccio.Ciò può essere evitato utilizzando inibitori della polimerasi che si dissociano dalla DNA polimerasi solo una volta raggiunta una determinata temperatura, da qui il termine PCR hot start.L'inibitore può essere un anticorpo che lega la polimerasi e la denatura alla temperatura di denaturazione iniziale (tipicamente 95°C).
Polimerasi ad alta fedeltà
Sebbene le DNA polimerasi amplificano in modo abbastanza accurato la sequenza stampo originale, possono verificarsi errori nell'abbinamento nucleotidico.Disadattamenti in applicazioni come la clonazione possono provocare trascrizioni troncate e proteine tradotte erroneamente o inattive a valle.Per evitare queste discrepanze, sono state identificate e incorporate nel flusso di lavoro polimerasi con attività di “correzione di bozze”.La prima polimerasi di correzione di bozze, Pfu, è stata identificata nel 1991 nel Pyrococcus furiosus.Questo enzima Pfu ha un'attività di esonucleasi da 3' a 5'.Quando il DNA viene amplificato, l'esonucleasi rimuove i nucleotidi non corrispondenti all'estremità 3' del filamento.Il nucleotide corretto viene quindi sostituito e la sintesi del DNA continua.L'identificazione di sequenze nucleotidiche errate si basa sull'affinità di legame del nucleoside trifosfato corretto con l'enzima, dove il legame inefficiente rallenta la sintesi e consente la corretta sostituzione.L'attività di correzione di bozze della polimerasi Pfu comporta meno errori nella sequenza finale rispetto alla Taq DNA polimerasi.Negli ultimi anni sono stati identificati altri enzimi di correzione di bozze e sono state apportate modifiche all'enzima Pfu originale per ridurre ulteriormente il tasso di errore durante l'amplificazione del DNA.
RT-PCR
La PCR a trascrizione inversa, o RT-PCR, consente l'uso dell'RNA come modello.Un ulteriore passaggio consente il rilevamento e l'amplificazione dell'RNA.L'RNA viene trascritto al contrario nel DNA complementare (cDNA), utilizzando la trascrittasi inversa.La qualità e la purezza del modello di RNA sono essenziali per il successo della RT-PCR.Il primo passo della RT-PCR è la sintesi di un ibrido DNA/RNA.La trascrittasi inversa ha anche una funzione RNasi H, che degrada la porzione di RNA dell'ibrido.La molecola di DNA a filamento singolo viene quindi completata dall'attività della DNA polimerasi DNA-dipendente della trascrittasi inversa in cDNA.L'efficienza della reazione del primo filamento può influenzare il processo di amplificazione.Da qui in poi, per amplificare il cDNA viene utilizzata la procedura PCR standard.La possibilità di convertire l'RNA in cDNA mediante RT-PCR presenta molti vantaggi e viene utilizzata principalmente per l'analisi dell'espressione genica.L'RNA è a filamento singolo e molto instabile, il che rende difficile lavorarci.Serve comunemente come primo passo nella qPCR, che quantifica le trascrizioni di RNA in un campione biologico.
qPCR e RT-qPCR
La PCR quantitativa (qPCR) viene utilizzata per rilevare, caratterizzare e quantificare gli acidi nucleici per numerose applicazioni.Nella RT-qPCR, le trascrizioni di RNA vengono spesso quantificate mediante trascrizione inversa prima in cDNA, come descritto sopra, e successivamente viene eseguita la qPCR.Come nella PCR standard, il DNA viene amplificato attraverso tre passaggi ripetitivi: denaturazione, ricottura e allungamento.Tuttavia, nella qPCR, la marcatura fluorescente consente la raccolta di dati man mano che la PCR procede.Questa tecnica presenta molti vantaggi grazie alla gamma di metodi e prodotti chimici disponibili.
Nella qPCR basata su colorante (tipicamente verde), la marcatura fluorescente consente la quantificazione delle molecole di DNA amplificate impiegando l'uso di un colorante legante il dsDNA.Durante ogni ciclo viene misurata la fluorescenza.Il segnale di fluorescenza aumenta proporzionalmente alla quantità di DNA replicato.Pertanto, il DNA viene quantificato in “tempo reale” (Fig. 3).Gli svantaggi della qPCR basata su colorante sono che è possibile esaminare un solo target alla volta e che il colorante si legherà a qualsiasi ds-DNA presente nel campione.
|
|
| Figura 3.Amplificazione di un modello di DNA mediante qPCR e misurazione del segnale di fluorescenza in tempo reale. |
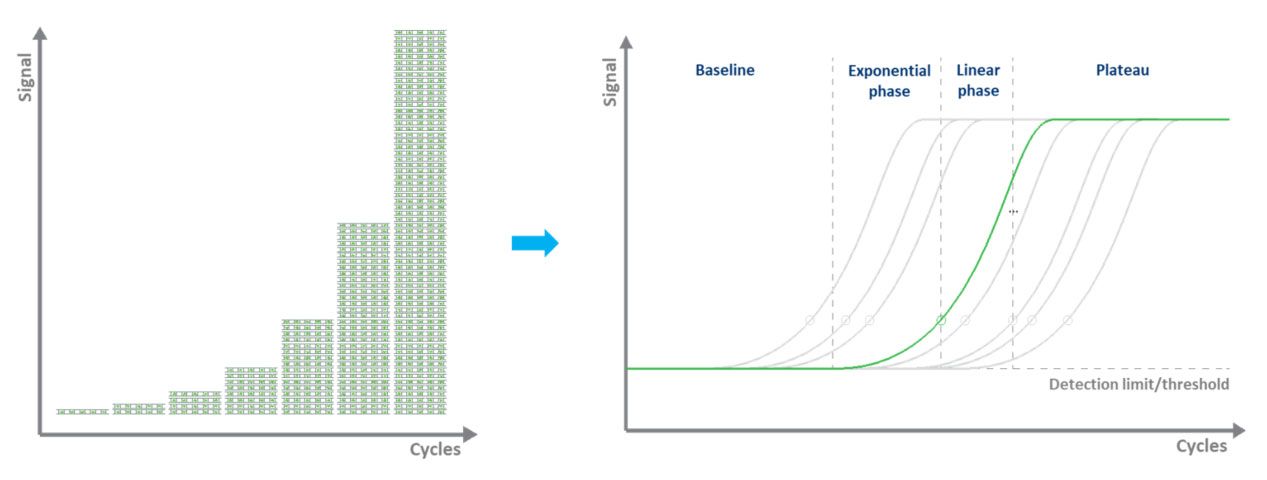
Nella qPCR basata su sonda, è possibile rilevare simultaneamente molti target in ciascun campione, ma ciò richiede l'ottimizzazione e la progettazione di sonde specifiche per il target utilizzate in aggiunta ai primer.Sono disponibili diversi tipi di design della sonda, ma il tipo più comune è una sonda per idrolisi, che incorpora un fluoroforo e un quencher.Il trasferimento di energia per risonanza di fluorescenza (FRET) impedisce l'emissione del fluoroforo tramite il quencher mentre la sonda è intatta.Tuttavia, durante la reazione PCR, la sonda viene idrolizzata durante l'estensione del primer e l'amplificazione della sequenza specifica a cui è legata.La scissione della sonda separa il fluoroforo dal quencher e determina un aumento della fluorescenza dipendente dall'amplificazione (Fig. 4).Pertanto, il segnale di fluorescenza proveniente da una reazione qPCR basata su sonda è proporzionale alla quantità di sequenza target della sonda presente nel campione.Poiché la qPCR basata su sonda è più specifica della qPCR basata su coloranti, è spesso la tecnologia utilizzata nei test diagnostici basati su qPCR.
| |
| Figura 4.Differenze tra qPCR basato su coloranti e basato su sonda. |
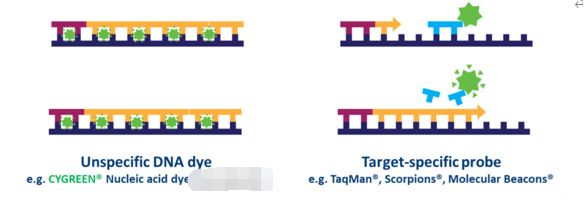
Amplificazione isotermica
Le tecniche PCR sopra menzionate richiedono costose apparecchiature di termociclaggio per aumentare e diminuire con precisione le temperature della camera per le fasi di denaturazione, ricottura ed estensione.Sono state sviluppate numerose tecniche che non necessitano di dispositivi così precisi e possono essere eseguite in un semplice bagnomaria o anche all'interno delle cellule di interesse.Queste tecniche sono chiamate collettivamente amplificazione isotermica e funzionano sulla base di un'amplificazione esponenziale, lineare o a cascata.
Il tipo più noto di amplificazione isotermica è l'amplificazione isotermica mediata da loop o LAMP.LAMP utilizza l'amplificazione esponenziale a 65 ⁰C per amplificare il DNA o l'RNA modello.Quando si esegue LAMP, da quattro a sei primer complementari alle regioni del DNA bersaglio vengono utilizzati con una DNA polimerasi per sintetizzare il nuovo DNA.Due di questi primer hanno sequenze complementari che riconoscono le sequenze negli altri primer e le legano, consentendo la formazione di una struttura ad “anello” nel DNA appena sintetizzato che poi aiuta la ricottura dei primer nei successivi cicli di amplificazione.La LAMP può essere visualizzata con diversi metodi, tra cui fluorescenza, elettroforesi su gel di agarosio o colorimetria.La facilità di visualizzare e rilevare la presenza o l'assenza del prodotto mediante colorimetria e la mancanza di attrezzature costose necessarie hanno reso LAMP un'opzione adatta per i test SARS-CoV-2 in aree in cui i test di laboratorio clinici non erano prontamente disponibili o per la conservazione e il trasporto dei campioni non era fattibile, o in laboratori che in precedenza non disponevano di apparecchiature per il termociclaggio PCR.
Orario di pubblicazione: 19 agosto 2023



