



RT-qPCR è sviluppato dalla normale tecnologia PCR.Aggiunge sostanze chimiche fluorescenti (coloranti fluorescenti o sonde fluorescenti) al sistema di reazione PCR tradizionale e rileva il processo di ricottura ed estensione della PCR in tempo reale in base ai loro diversi meccanismi luminescenti.Le variazioni del segnale fluorescente nel mezzo vengono utilizzate per calcolare la quantità di variazione del prodotto in ogni ciclo di PCR.Attualmente, i metodi più comuni sono il metodo del colorante fluorescente e il metodo della sonda.
Metodo colorante fluorescente:
Alcuni coloranti fluorescenti, come SYBR Green Ⅰ, PicoGreen, BEBO, ecc., non emettono luce da soli, ma emettono fluorescenza dopo essersi legati al solco minore del dsDNA.Pertanto, all'inizio della reazione PCR, la macchina non è in grado di rilevare il segnale fluorescente.Quando la reazione procede alla fase di estensione della ricottura (metodo in due fasi) o di estensione (metodo in tre fasi), i doppi filamenti vengono aperti in questo momento e la nuova DNA polimerasi Durante la sintesi del filamento, le molecole fluorescenti vengono combinate nel solco minore del dsDNA ed emettono fluorescenza.Con l'aumentare del numero di cicli di PCR, sempre più coloranti si combinano con dsDNA e anche il segnale fluorescente viene continuamente migliorato.Prendi SYBR Green Ⅰ come esempio.
Metodo della sonda:
La sonda Taqman è la sonda per idrolisi più comunemente utilizzata.C'è un gruppo fluorescente all'estremità 5′ della sonda, solitamente FAM.La sonda stessa è una sequenza complementare al gene bersaglio.C'è un gruppo di spegnimento fluorescente all'estremità 3 'del fluoroforo.Secondo il principio del trasferimento di energia di risonanza della fluorescenza (trasferimento di energia di risonanza di Förster, FRET), quando il gruppo fluorescente reporter (molecola fluorescente donatrice) e il gruppo fluorescente di estinzione (molecola fluorescente accettore)Pertanto, all'inizio della reazione PCR, quando la sonda è libera e intatta nel sistema, il gruppo fluorescente reporter non emetterà fluorescenza.Durante la ricottura, il primer e la sonda si legano al modello.Durante la fase di estensione, la polimerasi sintetizza continuamente nuove catene.La DNA polimerasi ha attività esonucleasica 5′-3′.Quando raggiunge la sonda, la DNA polimerasi idrolizzerà la sonda dallo stampo, separerà il gruppo fluorescente reporter dal gruppo fluorescente quencher e rilascerà il segnale fluorescente.Poiché esiste una relazione uno a uno tra la sonda e il templato, il metodo della sonda è superiore al metodo del colorante in termini di accuratezza e sensibilità del test.
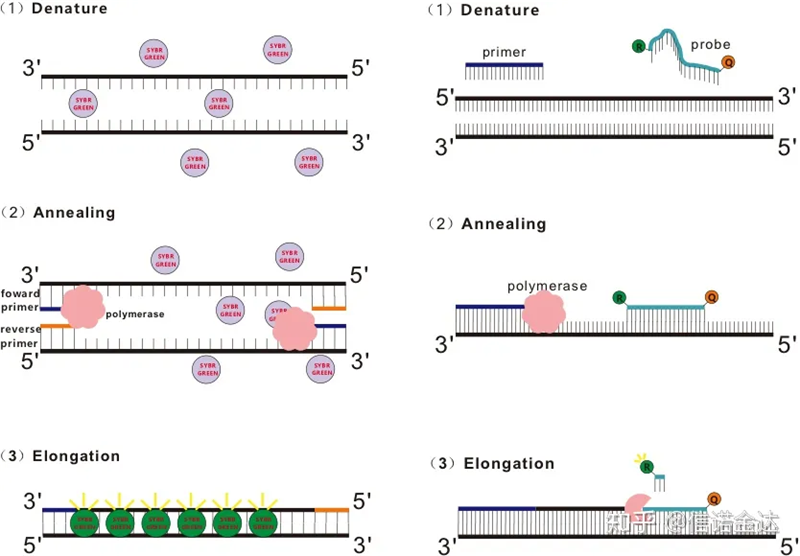
Fig 1 Principio di qRT-PCR
Disegno dell'innesco
I principi:
I primer dovrebbero essere progettati nella regione conservata della serie di acidi nucleici e avere specificità.
È meglio usare la sequenza del cDNA e anche la sequenza dell'mRNA è accettabile.In caso contrario, scopri il design della regione cds della sequenza del DNA.
La lunghezza del prodotto quantitativo fluorescente è di 80-150 bp, la più lunga è di 300 bp, la lunghezza del primer è generalmente compresa tra 17-25 basi e la differenza tra i primer a monte e a valle non dovrebbe essere troppo grande.
Il contenuto di G+C è compreso tra il 40% e il 60% e il 45-55% è il migliore.
Il valore TM è compreso tra 58 e 62 gradi.
Cercare di evitare primer dimeri e autodimeri, (non compaiono più di 4 paia di basi complementari consecutive) struttura a forcina, se inevitabile, creare ΔG<4.5kJ/mol* -
specifica L'omologia della sequenza eterogeneamente amplificata è preferibilmente inferiore al 70% o ha un'omologia di 8 basi complementari.
Banca dati:
Ricerca CottonFGD per parole chiave
Disegno dell'innesco:
Design del primer IDT-qPCR
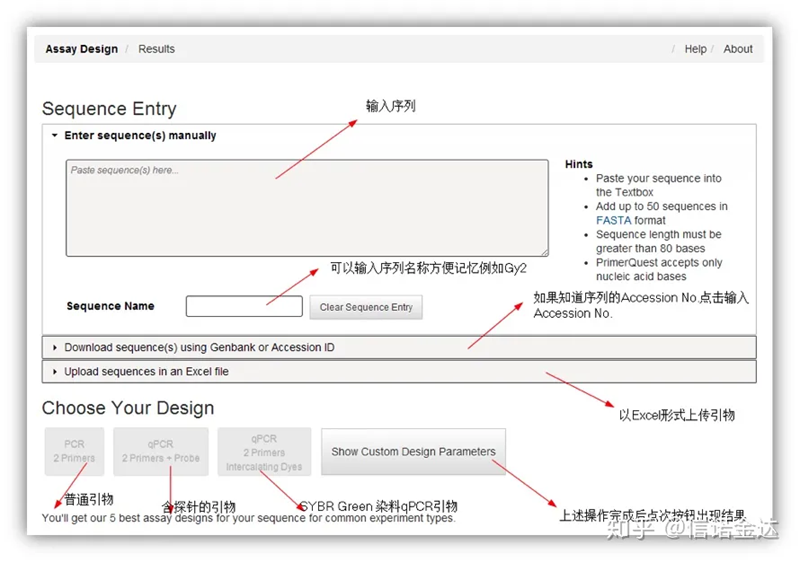
Fig2 Pagina dello strumento di progettazione del primer online IDT
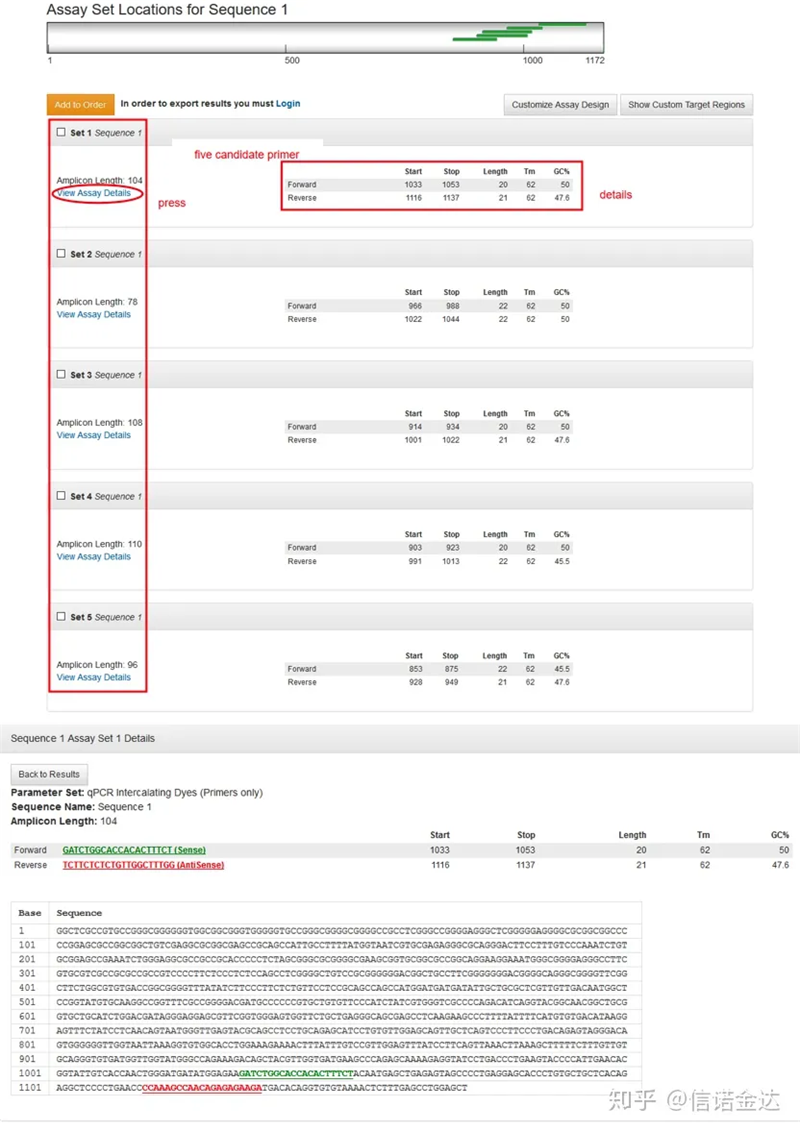
Fig3 visualizzazione della pagina dei risultati
Progettazione di primer lncRNA:
lncRNA:gli stessi passaggi dell'mRNA.
miRNA:Il principio del metodo stem-loop: poiché tutti i miRNA sono brevi sequenze di circa 23 nt, non è possibile eseguire il rilevamento diretto della PCR, quindi viene utilizzato lo strumento di sequenza stem-loop.La sequenza stem-loop è un DNA a filamento singolo di circa 50 nt, che può formare da solo una struttura a forcina.3 'L'estremità può essere progettata come una sequenza complementare al frammento parziale del miRNA, quindi il miRNA target può essere collegato alla sequenza stem-loop durante la trascrizione inversa e la lunghezza totale può raggiungere 70 bp, che è in linea con la lunghezza del prodotto amplificato determinato da qPCR.Progettazione del primer miRNA di coda .
Rilevamento specifico dell'amplificazione:
Database di esplosioni in linea: esplosione di CottonFGD per somiglianza di sequenza
Local blast: fare riferimento all'utilizzo di Blast+ per eseguire local blast, linux e macos possono stabilire direttamente un database locale, il sistema win10 può essere eseguito anche dopo l'installazione di ubuntu bash.Creare un database di esplosioni locali e un'esplosione locale;apri ubuntu bash su win10
Avviso: il cotone di montagna e il cotone delle isole marine sono colture tetraploidi, quindi il risultato dell'esplosione sarà spesso due o più corrispondenze.In passato, è probabile che l'utilizzo di NAU cds come database per eseguire l'esplosione trovasse due geni omologhi con solo poche differenze SNP.Di solito, i due geni omologhi non possono essere separati dal design del primer, quindi vengono trattati come la stessa cosa.Se c'è un indel evidente, il primer è solitamente disegnato sull'indel, ma questo può portare alla struttura secondaria del primer. L'energia libera aumenta, portando a una diminuzione dell'efficienza di amplificazione, ma questo è inevitabile.
Rilevamento della struttura secondaria del primer:
Passi:apri l'oligo 7 → inserisci la sequenza del modello → chiudi la finestra secondaria → salva → individua il primer sul modello, premi ctrl+D per impostare la lunghezza del primer → analizza varie strutture secondarie, come il corpo di auto-dimerizzazione, l'eterodimero, la forcina, la mancata corrispondenza, ecc. Le ultime due immagini nella Figura 4 sono i risultati del test dei primer.Il risultato del primer anteriore è buono, non vi è alcuna struttura evidente di dimero e forcina, nessuna base complementare continua e il valore assoluto dell'energia libera è inferiore a 4,5, mentre il primer posteriore mostra continuo Le 6 basi sono complementari e l'energia libera è 8,8;inoltre, all'estremità 3′ compare un dimero più serio e compare un dimero di 4 basi consecutive.Sebbene l'energia libera non sia elevata, il 3 'dimero Chl può influenzare seriamente la specificità dell'amplificazione e l'efficienza dell'amplificazione.Inoltre, è necessario verificare la presenza di forcine, eterodimeri e disallineamenti.
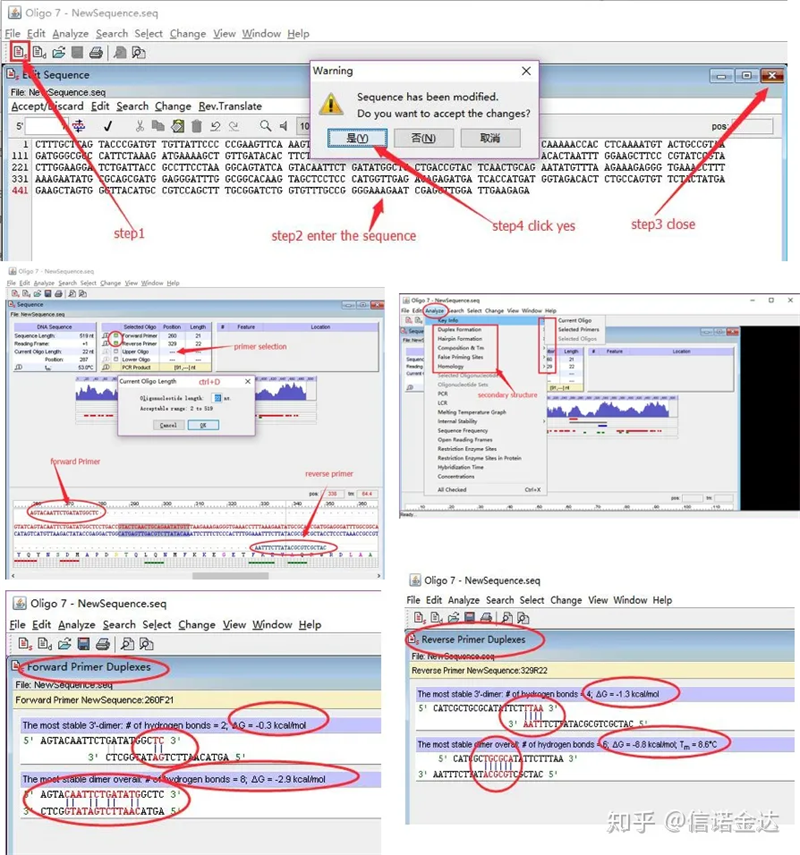
Risultati del rilevamento di Fig3 oligo7
Rilevamento dell'efficienza di amplificazione:
L'efficienza di amplificazione della reazione PCR influisce seriamente sui risultati della PCR.Anche in qRT-PCR, l'efficienza di amplificazione è particolarmente importante per i risultati quantitativi.Rimuovere altre sostanze, macchine e protocolli nel tampone di reazione.La qualità dei primer ha anche una grande influenza sull'efficienza di amplificazione di qRT-PCR.Per garantire l'accuratezza dei risultati, sia la quantificazione relativa della fluorescenza che la quantificazione assoluta della fluorescenza devono rilevare l'efficienza di amplificazione dei primer.È riconosciuto che l'effettiva efficienza di amplificazione qRT-PCR è compresa tra l'85% e il 115%.Ci sono due metodi:
1. Metodo della curva standard:
UN.Mescolare cDNA
B.Diluizione gradiente
c.qPCR
D.Equazione di regressione lineare per calcolare l'efficienza di amplificazione
2. LinRegPCR
LinRegPCR è un programma per l'analisi di dati RT-PCR in tempo reale, chiamati anche dati PCR quantitativi (qPCR) basati su SYBR Green o chimica simile.Il programma utilizza dati non corretti dalla linea di base, esegue una correzione della linea di base su ciascun campione separatamente, determina una finestra di linearità e quindi utilizza l'analisi di regressione lineare per adattare una linea retta attraverso il set di dati PCR.Dalla pendenza di questa linea viene calcolata l'efficienza PCR di ogni singolo campione.L'efficienza PCR media per amplicone e il valore Ct per campione vengono utilizzati per calcolare una concentrazione iniziale per campione, espressa in unità di fluorescenza arbitrarie.L'input e l'output dei dati avviene tramite un foglio di calcolo Excel.Solo campione
è richiesta la miscelazione, nessun gradiente
sono necessari passaggi:(Prendi Bole CFX96 come esempio, non proprio Machine con ABI chiaro)
sperimentare:è un esperimento qPCR standard.
Uscita dati qPCR:LinRegPCR è in grado di riconoscere due forme di file di output: RDML o quantificazione Risultato dell'amplificazione.Infatti, è il valore di rilevamento in tempo reale del numero di cicli e del segnale di fluorescenza da parte della macchina e l'amplificazione si ottiene analizzando il valore di variazione della fluorescenza dell'efficienza del segmento lineare.
Selezione dei dati: In teoria, il valore RDML dovrebbe essere utilizzabile.Si stima che il problema del mio computer sia che il software non è in grado di riconoscere RDML, quindi ho il valore di output excel come dati originali.Si consiglia di eseguire prima uno screening approssimativo dei dati, ad esempio l'errore di aggiunta di campioni, ecc. I punti possono essere eliminati nei dati di output (ovviamente non è possibile eliminarli, LinRegPCR ignorerà questi punti nella fase successiva)
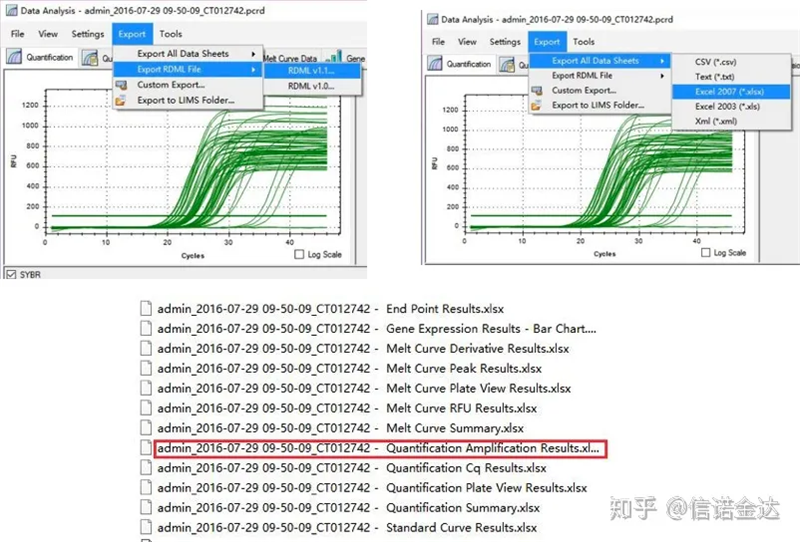
Fig5 Esportazione dati qPCR
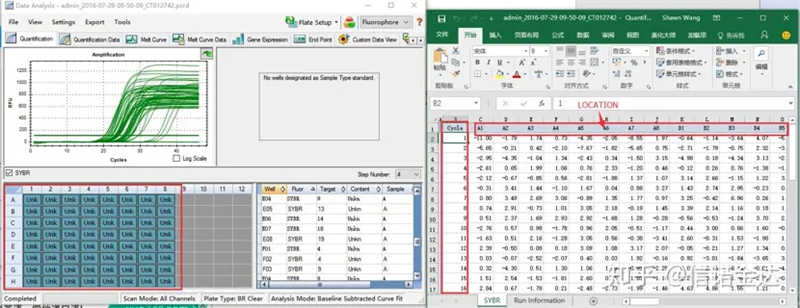
Fig6 selezione dei campioni candidati
Inserimento dati:Apri i risultati dell'amplificazione della qualificazione.xls, → apri LinRegPCR → file → leggi da excel → seleziona i parametri come mostrato nella Figura 7 → OK → fai clic su determina linee di base
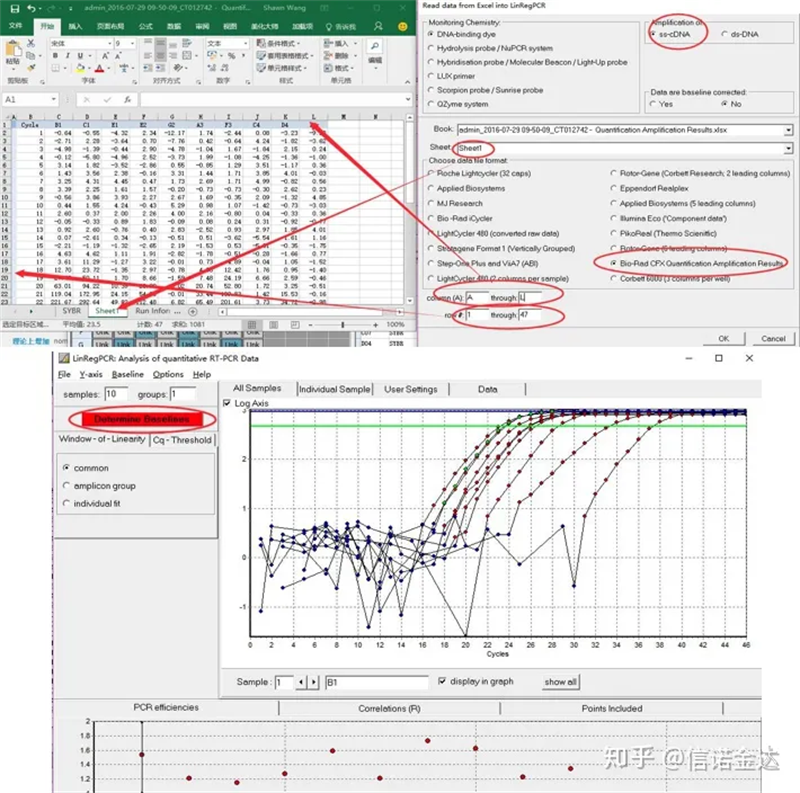
Fig7 fasi di input dei dati linRegPCR
Risultato:Se non c'è ripetizione, non è richiesto alcun raggruppamento.In caso di ripetizione, il raggruppamento può essere modificato nel raggruppamento del campione e il nome del gene viene inserito nell'identificatore, quindi lo stesso gene verrà automaticamente raggruppato.Infine, fai clic sul file, esporta Excel e visualizza i risultati.Verranno visualizzati l'efficienza di amplificazione e i risultati R2 di ciascun pozzetto.In secondo luogo, se si divide in gruppi, verrà visualizzata l'efficienza di amplificazione media corretta.Assicurarsi che l'efficienza di amplificazione di ciascun primer sia compresa tra l'85% e il 115%.Se è troppo grande o troppo piccolo, significa che l'efficienza di amplificazione del primer è scarsa.
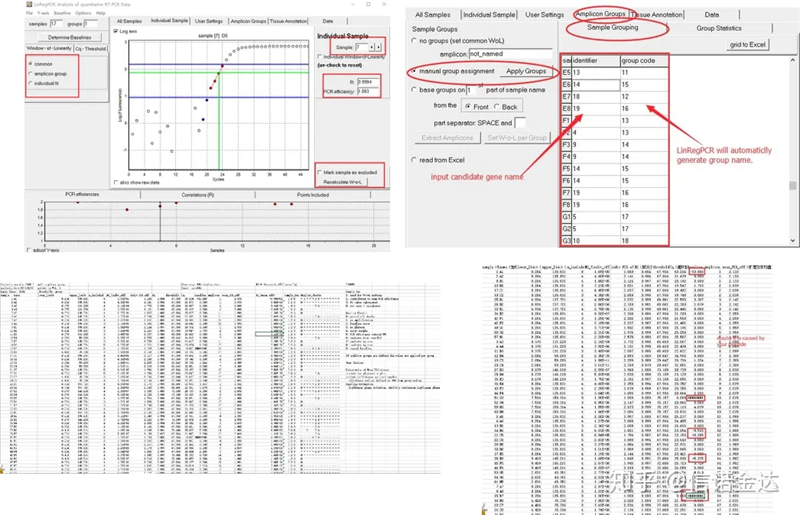
Fig 8 Risultato e output dei dati
Processo sperimentale:
Requisiti di qualità dell'RNA:
Purezza:1.72.0 indica che potrebbe esserci isotiocianato residuo.L'acido nucleico pulito A260/A230 dovrebbe essere intorno a 2. Se c'è un forte assorbimento a 230 nm, indica che ci sono composti organici come gli ioni fenato.Inoltre, può essere rilevato mediante elettroforesi su gel di agarosio all'1,5%.Punta il marcatore, perché lo ssRNA non ha denaturazione e il logaritmo del peso molecolare non ha una relazione lineare e il peso molecolare non può essere espresso correttamente.Concentrazione: Teoricamentenonmeno di 100ng/ul, se la concentrazione è troppo bassa, la purezza è generalmente bassa non alta
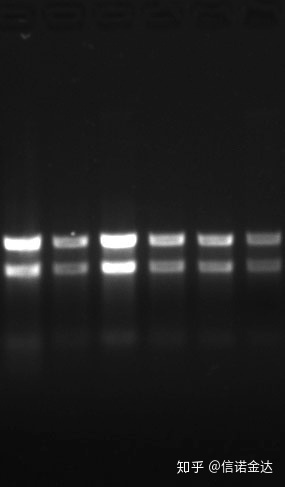
Gel di RNA Fig9
Inoltre, se il campione è prezioso e la concentrazione di RNA è elevata, si consiglia di aliquotarlo dopo l'estrazione e di diluire l'RNA a una concentrazione finale di 100-300 ng/ul per la trascrizione inversa.Inil processo di trascrizione inversa, quando l'mRNA viene trascritto, i primer oligo (dt) che possono legarsi specificamente alle code di poliA vengono utilizzati per la trascrizione inversa, mentre lncRNA e circRNA utilizzano primer esameri casuali (Random 6 mer) per la trascrizione inversa dell'RNA totale.Molte aziende hanno ora lanciato speciali kit di tailing.Per il metodo stem-loop, il metodo tailing è più conveniente, ad alto rendimento e risparmio di reagenti, ma l'effetto di distinguere i miRNA della stessa famiglia non dovrebbe essere buono come il metodo stem-loop.Ogni kit di trascrizione inversa ha requisiti per la concentrazione di primer gene-specifici (stem-loop).Il riferimento interno utilizzato per il miRNA è U6.Nel processo di inversione stem-loop, un tubo di U6 dovrebbe essere invertito separatamente e gli inneschi anteriore e posteriore di U6 dovrebbero essere aggiunti direttamente.Sia circRNA che lncRNA possono utilizzare HKG come riferimento interno.Inrilevamento del cDNA,
se non ci sono problemi con l'RNA, anche il cDNA dovrebbe andare bene.Tuttavia, se si persegue la perfezione dell'esperimento, è meglio utilizzare un gene di riferimento interno (Gene di riferimento, RG) in grado di distinguere gDNA da cds.Generalmente, RG è un gene di pulizia., HKG) come mostrato in Figura 10;A quel tempo, stavo producendo proteine di riserva di soia e usavo actina7 contenente introni come riferimento interno.La dimensione del frammento amplificato di questo primer nel gDNA era di 452 bp, e se il cDNA veniva usato come templato, era di 142 bp.Quindi i risultati del test hanno rilevato che una parte del cDNA era effettivamente contaminata dal gDNA e ha anche dimostrato che non vi erano problemi con il risultato della trascrizione inversa e che poteva essere utilizzato come modello per la PCR.È inutile eseguire l'elettroforesi su gel di agarosio direttamente con cDNA, ed è una banda diffusa, che non è convincente.
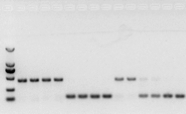
Figura 10 Rilevamento del cDNA
La determinazione delle condizioni qPCRnon è generalmente un problema secondo il protocollo del kit, principalmente nella fase del valore tm.Se alcuni primer non sono ben progettati durante la progettazione dei primer, determinando una grande differenza tra il valore tm e i 60°C teorici, si consiglia di miscelare il cDNA Dopo che i campioni sono stati miscelati, eseguire una PCR in gradiente con i primer e cercare di evitare di impostare la temperatura senza bande come valore TM.
Analisi dei dati
Il metodo di elaborazione PCR quantitativa della fluorescenza relativa convenzionale è fondamentalmente secondo 2-ΔΔCT.Modello per il trattamento dei dati.
Prodotti correlati:
PCR in tempo reale FacileTM –Taqman
PCR in tempo reale FacileTM –SYBR VERDE I
RT Easy I (Master Premix per la sintesi del cDNA del primo filamento)
RT Easy II (Master Premix per la sintesi del cDNA del primo filamento per qPCR)
Tempo di pubblicazione: 14 marzo 2023



