



1. Comprensione iniziale
In questa fase, dobbiamo comprendere alcuni concetti e terminologia, in modo da evitare di commettere errori davanti ai nostri anziani, come ad esempio:
D: Qual è la differenza tra RT-PCR, qPCR, PCR in tempo reale e RT-PCR in tempo reale?
Risposta: RT-PCR è PCR a trascrizione inversa(PCR a trascrizione inversa, RT-PCR), che è una variante ampiamente utilizzata della reazione a catena della polimerasi (PCR).In RT-PCR, un filamento di RNA viene retrotrascritto in DNA complementare, che viene quindi utilizzato come modello per l'amplificazione del DNA mediante PCR.
PCR in tempo reale e qPCR(Quantitative Rea-ltime-PCR) sono la stessa cosa, entrambi sono PCR quantitativa in tempo reale, il che significa che ogni ciclo di PCR ha record di dati in tempo reale, quindi il numero di modelli iniziali può essere regolato con un'analisi precisa.
Sebbene sia la PCR in tempo reale (PCR quantitativa fluorescente in tempo reale) che la PCR a trascrizione inversa (PCR a trascrizione inversa) sembrino essere abbreviate in RT-PCR, la convenzione internazionale è: RT-PCR si riferisce specificamente alla trascrizione inversaPCR , PCR in tempo reale è generalmente abbreviato in qPCR (PCR quantitativa in tempo reale).
E RT-PCR in tempo reale (RT-qPCR), è la PCR a trascrizione inversa combinata con la tecnologia quantitativa fluorescente: ottenere prima il cDNA (RT) dalla trascrizione inversa dell'RNA, quindi utilizzare la PCR in tempo reale per l'analisi quantitativa (qPCR).La maggior parte dei laboratori fa RT-qPCR, cioè ricerca sulla down-regulation dell'espressione dell'RNA, quindi il qPCR di cui tutti parlano in laboratorio si riferisce in realtà a RT-qPCR, ma non dimenticare che ci sono ancora molti test del DNA nelle applicazioni cliniche.Analisi quantitativa, come il rilevamento dell'HBV del virus dell'epatite B.
Domanda: Dopo aver letto un sacco di PCR quantitativa fluorescente, perché il frammento amplificato dovrebbe essere controllato nell'intervallo di 80-300 bp?
Risposta: La lunghezza di ciascuna sequenza genica è diversa, alcune sono diversi kb, alcune sono centinaia di bp, ma abbiamo solo bisogno di richiedere che la lunghezza del prodotto sia 80-300 bp quando si progettano primer, troppo corti o troppo lunghi non sono adatti per il rilevamento PCR quantitativo fluorescente.Il frammento del prodotto è troppo corto per essere distinto dal primer-dimero.La lunghezza del primer-dimero è di circa 30-40 bp ed è difficile distinguere se si tratta di un primer-dimero o di un prodotto se è inferiore a 80 bp.Se il frammento del prodotto è troppo lungo, superiore a 300 bp, porterà facilmente a una bassa efficienza di amplificazione e non sarà in grado di rilevare efficacemente la quantità del gene.
Ad esempio, quando conti quante persone ci sono in una classe, devi solo contare quante bocche ci sono.Lo stesso vale quando rilevi i geni, devi solo rilevare una certa sequenza di un gene per rappresentare L'intera sequenza andrà bene.Se vuoi contare le persone, devi contare sia la bocca che il naso, le orecchie e gli occhiali, ed è facile sbagliare.
Per espandere, nella ricerca biologica, ci sono molti casi di ricerca da un punto all'altro, poiché la sequenza genica di qualsiasi specie è molto lunga, non è necessario e impossibile misurare tutti i frammenti, come il sequenziamento batterico 16S, che consiste nell'eseguire la sequenza conservativa dei saggi batterici per dedurre il numero di una determinata popolazione di batteri.
D: Qual è la lunghezza ottimale per il design del primer qPCR?
Risposta: In generale, la lunghezza del primer è di circa 20-24 bp, che è migliore.Naturalmente, dobbiamo prestare attenzione al valore TM del primer durante la progettazione del primer, poiché questo è correlato alla temperatura di ricottura ottimale.Dopo molti esperimenti, è stato dimostrato che 60°C è un valore TM migliore.Se la temperatura di ricottura è troppo bassa, porterà facilmente ad un'amplificazione non specifica.Se la temperatura di ricottura è troppo alta, l'efficienza di amplificazione sarà relativamente bassa, il picco della curva di amplificazione inizierà più tardi e il valore CT sarà ritardato.
D: In che modo il metodo del colorante è diverso dal metodo della sonda?
Risposta: metodo coloranteAlcuni coloranti fluorescenti, come SYBR Green Ⅰ, PicoGreen, BEBO, ecc., non emettono luce da soli, ma emettono fluorescenza dopo essersi legati al solco minore del DNA a doppio filamento.Pertanto, all'inizio della reazione PCR, la macchina non è in grado di rilevare il segnale fluorescente.Quando la reazione raggiunge la fase di ricottura-estensione, il doppio filamento viene aperto e un nuovo filamento viene sintetizzato sotto l'azione della DNA polimerasi e la molecola fluorescente si lega al solco minore del dsDNA.Con l'aumentare del numero di cicli di PCR, sempre più coloranti vengono combinati con il DNA a doppio filamento e anche il segnale fluorescente viene continuamente migliorato.Il metodo della tintura è utilizzato principalmente nella ricerca scientifica.
PS: Fai attenzione quando fai l'esperimento, il colorante deve essere combinato con il DNA umano, fai attenzione a trasformarlo in una persona fluorescente.
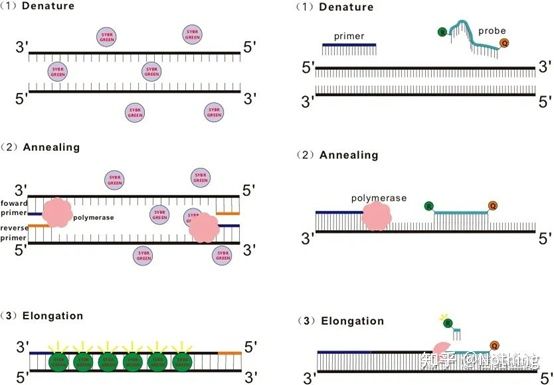
Metodo colorante (sinistra) Metodo sonda (destra)
PS: Fai attenzione quando fai l'esperimento, il colorante deve essere combinato con il DNA umano, fai attenzione a trasformarlo in una persona fluorescente.
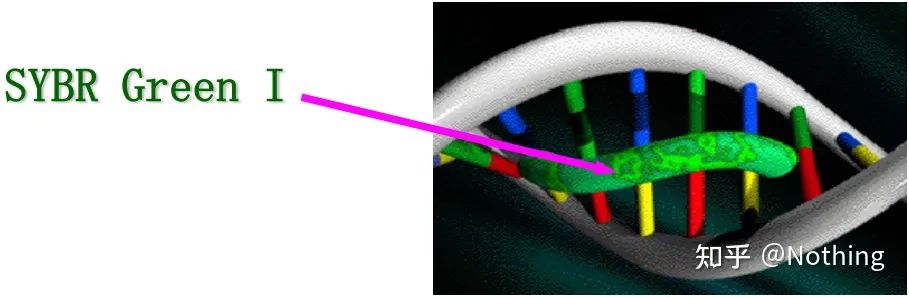
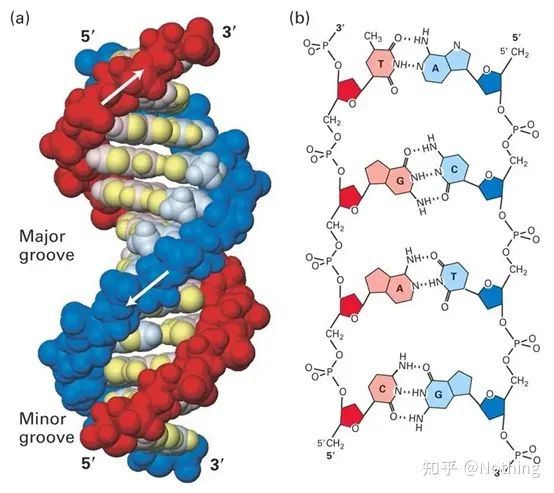
SYBR Green Ⅰ si lega al solco minore del DNA
Metodo sondaLa sonda Taqman è la sonda per idrolisi più comunemente utilizzata.C'è un gruppo fluorescente all'estremità 5' della sonda, solitamente FAM, e la sonda stessa è una sequenza complementare al gene bersaglio.C'è un gruppo di spegnimento fluorescente all'estremità 3 '.Secondo il principio del trasferimento di energia di risonanza della fluorescenza (trasferimento di energia di risonanza di Förster, FRET), quando il gruppo fluorescente del reporter (molecola fluorescente del donatore) e il gruppo fluorescente dell'estinzione (molecola fluorescente dell'accettore) sono eccitati.Pertanto, all'inizio della reazione PCR, quando la sonda è libera e intatta nel sistema, il gruppo fluorescente reporter non emetterà fluorescenza.Durante la ricottura, il primer e la sonda si legano al modello.Durante la fase di estensione, la polimerasi sintetizza continuamente nuove catene.La DNA polimerasi ha attività esonucleasica 5′-3′.Quando raggiunge la sonda, la DNA polimerasi idrolizzerà la sonda dallo stampo, separerà il gruppo fluorescente reporter dal gruppo fluorescente quencher e rilascerà il segnale fluorescente.Poiché esiste una relazione uno a uno tra la sonda e il templato, il metodo della sonda è superiore al metodo del colorante in termini di accuratezza e sensibilità del test.Il metodo della sonda è utilizzato principalmente nella diagnosi.
D: Cos'è la quantificazione assoluta?Cos'è la quantificazione relativa?
Risposta: La quantificazione assoluta si riferisce al calcolo del numero di copie iniziale del campione da testare mediante qPCR, ad esempio quanti virus HBV sono presenti in 1 ml di sangue.Il risultato ottenuto dalla quantificazione relativa è la variazione della quantità del gene bersaglio in un campione specifico rispetto a un altro campione di riferimento e l'espressione genica è sovraregolata o sottoregolata.
D: La quantità di estrazione dell'RNA, l'efficienza della trascrizione inversa e l'efficienza dell'amplificazione influenzeranno i risultati sperimentali?
D: La conservazione dei campioni, i reagenti di estrazione, i reagenti di trascrizione inversa e i materiali di consumo che trasmettono la luce influiranno sui risultati sperimentali?
D: Quale metodo può correggere i dati sperimentali?
Per quanto riguarda questi problemi, li descriveremo in dettaglio nelle sezioni avanzate e avanzate di seguito.
2. Conoscenza avanzata
Per quanto riguarda la PCR quantitativa fluorescente in tempo reale, dobbiamo riconoscere la realtà che ogni anno vengono pubblicati migliaia di articoli di ricerca scientifica, tra cui la tecnologia PCR quantitativa fluorescente non è un piccolo numero.
Se non esiste uno standard comune per misurare l'esperimento PCR quantitativo fluorescente, i risultati possono variare notevolmente.Per lo stesso gene della stessa specie, con lo stesso metodo di elaborazione, anche i risultati del rilevamento varieranno ampiamente e sarà difficile per i ritardatari ripetere gli stessi risultati.Nessuno sa cosa è giusto e cosa è sbagliato.
Questo significa che la PCR quantitativa fluorescente è una tecnologia cheat o una tecnologia inaffidabile?No, è perché la PCR quantitativa fluorescente è più sensibile e più accurata, e un'operazione leggermente sbagliata produrrà risultati completamente opposti.Una piccola perdita è lontana mille miglia.L'autore dell'articolo può essere ripetutamente torturato dai revisori.Allo stesso tempo, anche i revisori della rivista hanno difficoltà a scegliere tra diversi risultati sperimentali.
Tutto sommato, indicando una mancanza di consenso negli esperimenti di PCR in tempo reale.A tal fine, scienziati di alto livello del settore hanno iniziato a formulare standard,richiedere ai contributori di fornire nell'articolo alcuni dettagli sperimentali e di elaborazione dei dati necessari (inclusi i dati necessari) per soddisfare questi standard .
I revisori possono giudicare la qualità dell'esperimento leggendo questi dettagli;i futuri lettori possono anche usarlo per ripetere l'esperimento o migliorarlo.Quindi i risultati sperimentali così ottenuti sono ricchi di informazioni, di alta qualità e fruibili.
MIBBI (Informazioni Minime per Indagini Biologiche e Biomediche -http://www.mibbi.org) venuto in essere.MIBBI è un progetto che fornisce standard per gli esperimenti.È pubblicato in natura.Questo progetto è rivolto a vari esperimenti biologici, tra cui biologia cellulare, Microarray, qPCR di cui parleremo ora, ecc., e prevede per ogni tipo di esperimento al momento della presentazione dei manoscritti.Tali informazioni dovrebbero essere fornite in ogni momento.
Nel progetto MIBBI, ci sono due articoli relativi alla PCR quantitativa fluorescente, vale a dire:
·RDML (Real-Time PCR Data Markup Language) – un linguaggio strutturato e una guida di reporting per dati PCR quantitativi in tempo reale;
·MIQE (Informazioni minime per la pubblicazione di esperimenti di PCR quantitativa in tempo reale) – informazioni minime per la pubblicazione di articoli sugli esperimenti di PCR quantitativa in tempo reale.
Innanzitutto, parliamo di RDML, la specifica terminologica.
Se non esiste una definizione standard per tutto, è impossibile continuare la discussione, motivo per cui la spiegazione dei termini è così importante nell'esame.
La terminologia utilizzata nell'esperimento PCR quantitativa fluorescente include il seguente contenuto.QIAGEN ha fatto il miglior riassunto per noi.I seguenti sono tutti asciuttimerce .
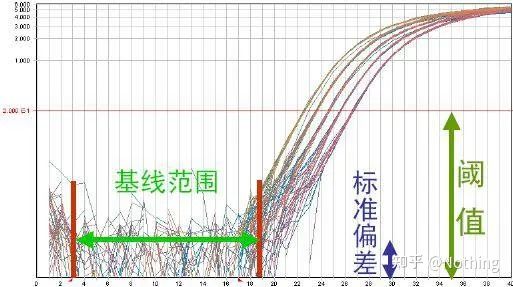
Curva di amplificazione
La curva di amplificazione si riferisce alla curva realizzata durante il processo PCR, con il numero del ciclo come ascissa e l'intensità di fluorescenza in tempo reale durante la reazione come ordinata.
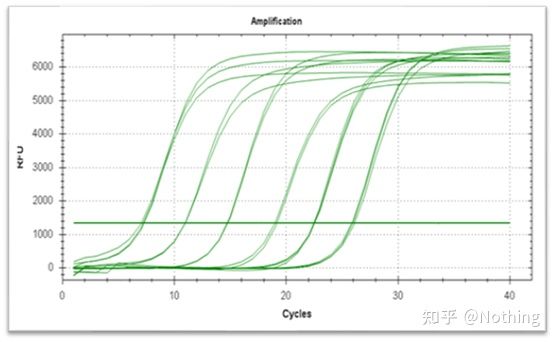
Un'ottima curva di amplificazione dovrebbe avere le seguenti caratteristiche: la linea di base è piatta o leggermente diminuita e non vi è alcuna evidente tendenza al rialzo;il punto di flesso della curva è chiaro e la pendenza della fase esponenziale è proporzionale all'efficienza di amplificazione.Maggiore è la pendenza, maggiore è l'efficienza di amplificazione;la curva di amplificazione complessiva Il parallelismo è buono, indicando che l'efficienza di amplificazione di ciascun tubo è simile;la fase esponenziale della curva di amplificazione dei campioni a bassa concentrazione è evidente.
Linea di base (linea di base)
La linea di base è il livello di rumore del primo ciclo, solitamente misurato tra il 3° e il 15° ciclo, poiché l'aumento del valore di fluorescenza causato dal prodotto di amplificazione non può essere rilevato durante questo periodo.Il numero di cicli utilizzati per calcolare la linea di base può essere variato e potrebbe essere necessario ridurlo se si utilizzano quantità elevate di templato o se il livello di espressione del gene target è elevato.
L'impostazione della linea di base richiede la visualizzazione dei dati di fluorescenza dalla curva di amplificazione della linearità.La linea di base è impostata in modo che la crescita della curva di amplificazione inizi con un numero di cicli maggiore del numero massimo del ciclo di base.Le linee di base devono essere impostate individualmente per ciascuna sequenza target.I valori medi di fluorescenza rilevati nei primi cicli devono essere sottratti dai valori di fluorescenza ottenuti nei prodotti amplificati.Le versioni più recenti di vari software per PCR in tempo reale consentono l'ottimizzazione automatica delle impostazioni della linea di base per i singoli campioni.
Durante i primi cicli della reazione di amplificazione PCR, il segnale di fluorescenza non cambia molto.L'avvicinamento a una linea retta è chiamato linea di base, ma se osserviamo attentamente i primi cicli, vediamo che all'interno della linea di base c'è ciò che sta accadendo nell'immagine qui sotto.
Sfondo Sfondo si riferisce a
il valore di fluorescenza non specifico nella reazione .Ad esempio: spegnimento della fluorescenza inefficiente;o un gran numero di modelli di DNA a doppio filamento dovuti all'uso di SYBR Green.Le componenti di fondo del segnale vengono rimosse matematicamente dall'algoritmo del software Real-Time PCR.
Segnale del giornalista
Il segnale del reporter si riferisce al segnale fluorescente generato da SYBR Green o da sonde specifiche della sequenza etichettate in modo fluorescente durante la PCR in tempo reale.
Segnale reporter normalizzato (RN)
RN si riferisce all'intensità di fluorescenza del colorante reporter divisa per l'intensità di fluorescenza del colorante di riferimento passivo misurata ad ogni ciclo.
Colorante di riferimento passivo
In alcune PCR in tempo reale,il colorante fluorescente ROX viene utilizzato come riferimento interno per normalizzare il segnale fluorescente.Corregge le variazioni dovute a pipettaggio impreciso, posizione del pozzetto e fluttuazioni di fluorescenza pozzetto per pozzetto.

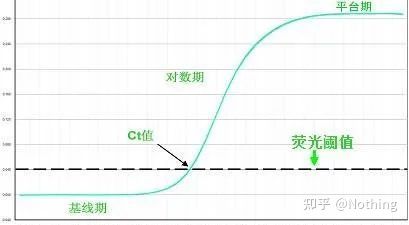
La soglia di fluorescenza (soglia)
è stato regolato al di sopra del valore di fondo e significativamente al di sotto del valore di plateau della curva di amplificazione.Deve trovarsi nella regione lineare della curva di amplificazione, che rappresenta l'intervallo log-lineare del rilevamento della PCR.Le soglie devono essere impostate nella visualizzazione della curva di amplificazione logaritmica in modo che la fase lineare logaritmica della PCR sia facilmente identificabile.Se sono presenti più geni target nella PCR in tempo reale, la soglia deve essere impostata per ciascun target .Generalmente, il segnale di fluorescenza dei primi 15 cicli di reazione PCR viene utilizzato come segnale di fondo della fluorescenza e la soglia di fluorescenza è 10 volte la deviazione standard del segnale di fluorescenza dei primi 3-15 cicli di PCR e la soglia di fluorescenza viene impostata nella fase esponenziale dell'amplificazione PCR.In generale, ogni strumento ha la sua soglia di fluorescenza impostata prima dell'uso.
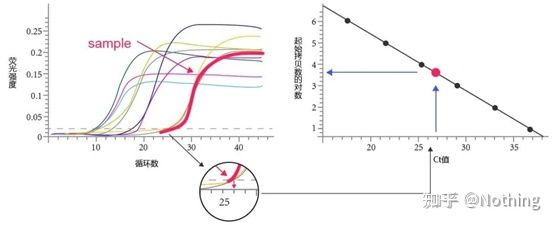
Soglia del ciclo (CT) o punto di attraversamento (CP)
Il ciclo in cui la curva di amplificazione attraversa la soglia (ovvero il punto in cui il rilevamento della fluorescenza aumenta in modo significativo).CT può essere una frazione e può essere calcolata la quantità di modello iniziale.Il valore CT rappresenta il numero di cicli sperimentati quando il segnale fluorescente in ciascuna provetta di reazione PCR raggiunge la soglia impostata.Esiste una relazione lineare tra il valore CT di ciascun modello e il logaritmo del numero di copie iniziale del modello, ilmaggiore è il numero di copie iniziali, minore è il valore CT e viceversa.Una curva standard può essere realizzata utilizzando uno standard con numero di copie iniziale noto, in cui l'ascissa rappresenta il valore CT e l'ordinata rappresenta il logaritmo del numero di copie iniziale.Pertanto, fintanto che si ottiene il valore CT del campione sconosciuto, il numero di copie iniziale del campione può essere calcolato dalla curva standard.
Valore ΔCT
Il valore ΔCT descrivela differenza tra il gene target e il corrispondente valore CT del gene endogeno di riferimento, come un gene di pulizia, e viene utilizzato per normalizzare la quantità di modello utilizzato:
⇒ΔCT = CT (gene bersaglio) – CT (gene di riferimento endogeno)
Valore ΔΔCT
Il valore ΔΔCT descrive la differenza tra il valore medio ΔΔCT di un campione di interesse (ad esempio, cellule stimolate) e il valore medio ΔΔCT di un campione di riferimento (ad esempio, cellule non stimolate).Il campione di riferimento è anche chiamato campione di calibrazione e tutti gli altri campioni sono normalizzati a questo per la quantificazione relativa:
⇒ΔΔCT = ΔCT medio (campione di interesse) – ΔCT medio (campione di riferimento)
Geni di riferimento endogeni (geni di riferimento endogeni)
I livelli di espressione dei geni di riferimento endogeni, come i geni domestici (geni domestici), non differiscono tra i campioni.Il confronto dei valori CT del gene di riferimento con il gene target consente di normalizzare il livello di espressione del gene target alla quantità di input RNA o cDNA (vedere la sezione sui valori ΔCT sopra).
Geni di riferimento interni corretti perpossibile degradazione dell'RNA o presenza di inibitori enzimatici nei campioni di RNA, nonché variazioni nel contenuto di RNA, efficienza di trascrizione inversa, recupero dell'acido nucleico e manipolazione del campione.Per selezionare i geni di riferimento ottimali, abbiamo modificato l'algoritmo per consentire la selezione del riferimento ottimale in base all'impostazione sperimentale.
Controllo interno
Una sequenza di controllo che viene amplificata nella stessa reazione della sequenza target e sondata con una sonda diversa (ad esempio, eseguendo la PCR duplex).I controlli interni vengono spesso utilizzati per escludere amplificazioni fallite, ad esempio quando la sequenza target non viene rilevata.
Campione di calibrazione
Un campione di riferimento (ad esempio, RNA purificato da una linea cellulare o da un tessuto) utilizzato nella quantificazione relativa per confrontare tutti gli altri campioni per determinare il livello di espressione relativo di un gene.Il campione di calibrazione può essere qualsiasi campione, ma di solito è un controllo (ad esempio, un campione non trattato o un campione dal tempo zero dell'esperimento).
Controlli positivi
utilizzare le reazioni di controllo conquantità note di modello.I controlli positivi vengono spesso utilizzati per verificare che un set di primer o un set di primer-sonda funzioni correttamente e che la reazione sia impostata correttamente.
Nessun controllo del modello (NTC)
Una reazione di controllo che contiene tutti i componenti necessari della reazione di amplificazione tranne lo stampo, che di solito viene sostituito con acqua.L'uso di NTC può trovare la contaminazione causata dalla contaminazione del reagente o da DNA estraneo, garantendo così l'autenticità e l'affidabilità dei dati di rilevamento.L'amplificazione del controllo NTC indica contaminazione.
Nessun controllo RT (NRT)
Il processo di estrazione dell'RNA può contenere DNA genomico residuo, che è estremamente dannoso ed è il colpevole che influisce sulla qualità dei dati e il nemico naturale di qPCR, quindi quando si progettano esperimenti, deve essere progettato per amplificare solo il rilevamento dell'RNA.Ci sono due modi, uno è progettare primer attraverso gli introni, l'altro è rimuovere completamente il DNA, qual è il migliore, che verrà discusso in seguito.Il controllo NTR è uno specchio magico per rilevare l'inquinamento del DNA.Se c'è amplificazione, significa che c'è inquinamento.
Standard
Gli standard sono campioni di concentrazione nota o numero di copie che vengono utilizzati per costruire una curva standard.Per garantire la stabilità dello standard, il frammento genico viene solitamente clonato nel plasmide e utilizzato come standard.
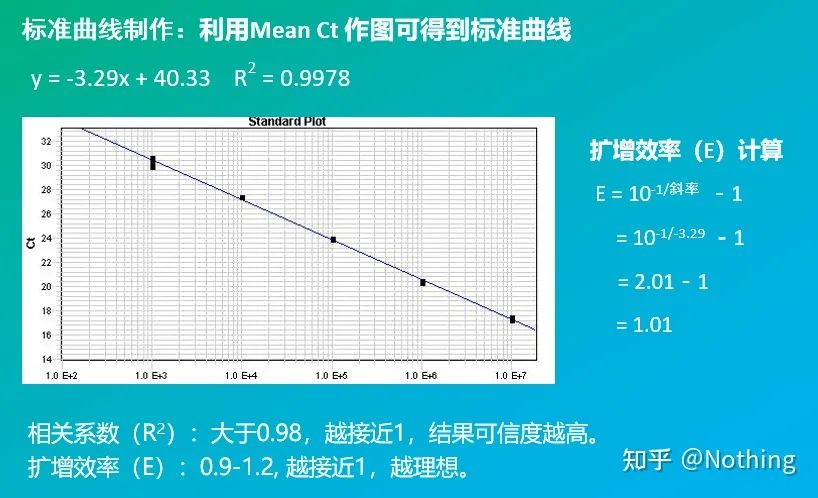
La curva standard
viene solitamente diluito in almeno 5 gradienti di concentrazione con il prodotto standard in base al rapporto di raddoppio e vengono disegnati 5 punti nelle coordinate del valore CT e del numero di copie e i punti sono collegati per formare una linea per generare una curva standard.Per ogni curva standard, è necessario verificarne la validità.Il valore della pendenza è compreso tra –3,3 e –3,8 e ciascuna concentrazione viene eseguita in triplicato.I punti che sono significativamente diversi da altri punti dovrebbero essere scartati.Il valore CT del campione da testare viene inserito nella curva standard ed è possibile calcolare il livello di espressione del campione da testare.
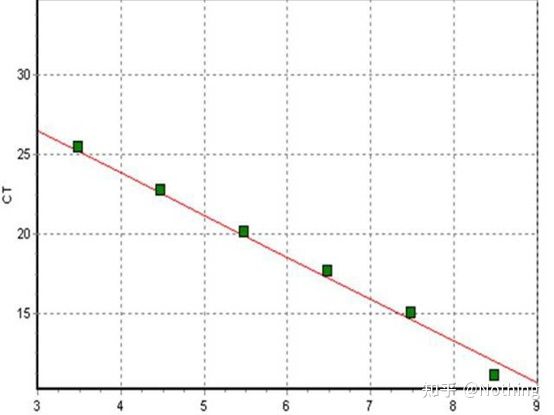
Il valore CT del campione da testare viene inserito nella curva standard ed è possibile calcolare il numero di copie iniziale del campione da testare.
Rendimento e pendenza
La pendenza della curva standard rappresenta l'efficienza della PCR in tempo reale.
·Una pendenza di -3,322 indica che l'efficienza di amplificazione della PCR è pari a 1, o efficiente al 100%, e che la quantità di prodotto della PCR raddoppia a ogni ciclo.
·Una pendenza inferiore a –3,322 (ad es. –3,8) indica un'efficienza della PCR
·Una pendenza maggiore di –3.322 (es. –3.0) indica che l'efficienza della PCR sembra essere maggiore del 100%, il che è curioso, come potrebbe un ciclo di PCR generare più del doppio del prodotto amplificato?Questa situazione si verifica nella fase non lineare della reazione PCR, cioè c'è una grande quantità di amplificazione non specifica.
curva di fusione
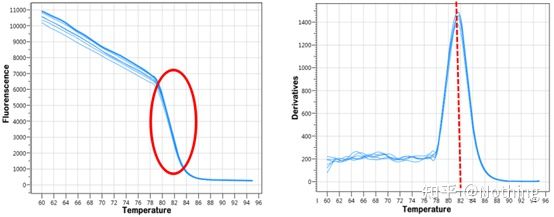
Dopo che l'amplificazione qPCR è stata completata, il prodotto PCR viene riscaldato.All'aumentare della temperatura, il prodotto di amplificazione a doppio filamento si scioglie gradualmente, determinando una diminuzione dell'intensità della fluorescenza.Quando viene raggiunta una certa temperatura (Tm), un gran numero di prodotti fonderà.La fluorescenza diminuisce bruscamente.Diversi prodotti PCR hanno diversi valori Tm e diverse temperature di fusione, in modo da poter identificare la specificità della PCR.
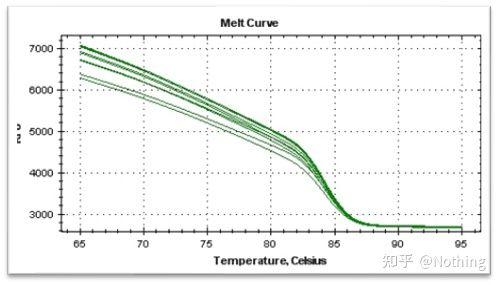
Curva di fusione (curva derivata)
La curva di fusione è derivata per formare una mappa dei picchi, che può visualizzare in modo più intuitivo la situazione dei frammenti di prodotto PCR.Poiché la temperatura di fusione è il valore Tm del frammento di DNA, è possibile giudicare alcuni parametri che influenzano il valore Tm del frammento di DNA, come la dimensione del frammento, il contenuto di GC, ecc. In generale, secondo i nostri principi di progettazione del primer,la lunghezza del prodotto amplificato è compresa tra 80 e 300 bp, quindi la temperatura di fusione dovrebbe essere compresa tra 80°C e 90°C.
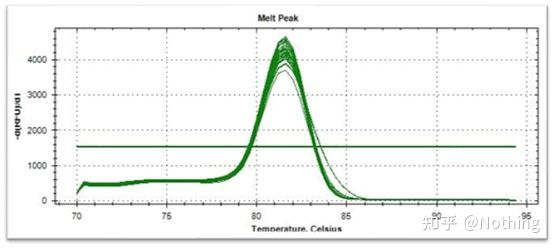
Interpretazione della curva di fusione: Se l'unico picco principale appare tra 80°C-90°C, significa che la PCR quantitativa fluorescente è perfetta;se il picco principale compare tra gli 80°C-90°C e i picchi vari compaiono al di sotto degli 80°C, si considera sostanzialmente il primer dimer.Puoi provare ad aumentare la temperatura di ricottura per risolverlo;se il picco principale appare tra 80°C-90°C, e il picco vario appare di nuovo quando la temperatura sale, si considera sostanzialmente che ci sia contaminazione del DNA, e il DNA deve essere rimosso nella fase iniziale dell'esperimento.
Naturalmente, ci sono ancora alcune situazioni anomale, che verranno analizzate una per una di seguito.
3. Conoscenza avanzata
Per fare qPCR, devo dire MIQE,Informazioni minimeper la pubblicazione diQuantitativoPCR in tempo realeEsperimenti: le informazioni minime per la pubblicazione di articoli sulla PCR quantitativa in tempo realeesperimenti.Per semplificare la comprensione di tutti, semplificheremo il contenuto chiave.
Puoi cercare il testo originale di MIQE su Internet e la cosa più importante è che stipuli illista di controllo dei dati che deve essere fornita quando si pubblica un articolo .
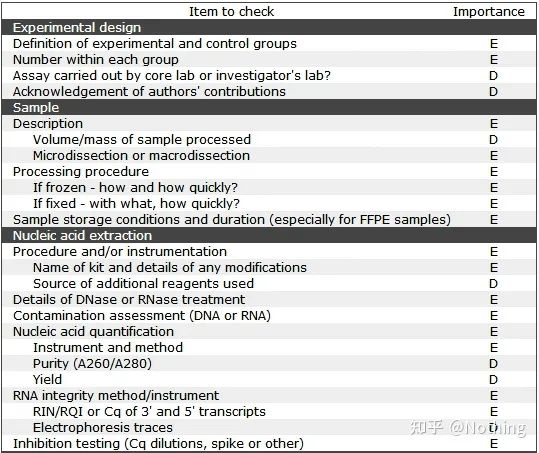
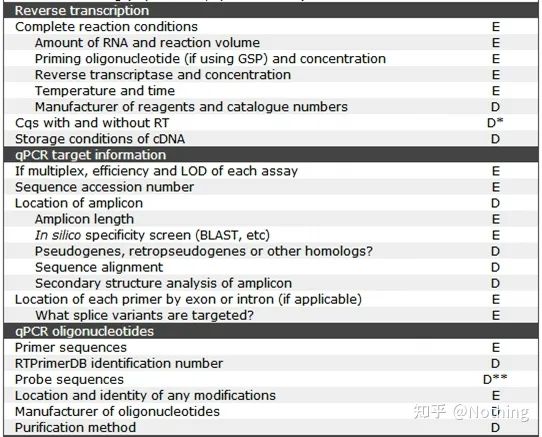
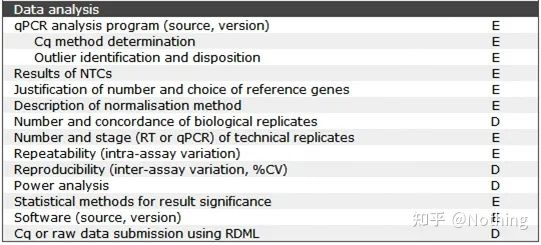
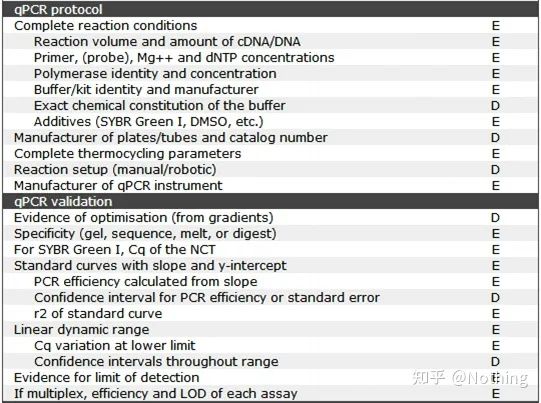
I revisori possono giudicare la qualità dell'esperimento leggendo questi dettagli;i futuri lettori possono anche usarlo per ripetere o migliorare l'esperimento.
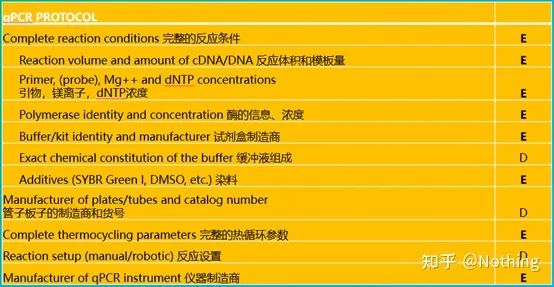
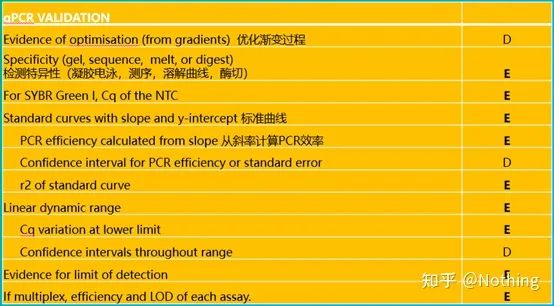
Vale la pena notare che in questo elenco l'importanza di ciascun elenco è contrassegnata rispettivamente con E o D.Cosa significa?E: informazioni essenziali (devono essere presentate);D: informazioni desiderabili (fornire il più possibile).
MIQE (1)—Progettazione sperimentale
Molti furfanti che hanno completato la loro difesa dopo aver terminato gli studi universitari non sapranno come progettare un esperimento in modo indipendente, aprire i loro quaderni e fare ciò che l'insegnante dice loro di fare.Di conseguenza, il design sperimentale non era rigoroso e la redazione della rivista ha detto che volevano inventare questa e quella foto, quindi l'hanno fatto in uno stato di stordimento.Ecco come sono fatti gli stronzi!

Più vicino a casa, il primo principio dell'esperimento è determinareil rigore della logica sperimentale.La cosa più fondamentale è il disegno sperimentale, e la cosa più importante del disegno sperimentale è come impostare il campione target, il campione di riferimento (controllo) e il numero di ripetizioni, in modo che i dati sperimentali possano essere referenziati, comparabili e convincenti.
Il campione bersagliosi riferisce al campione che ci richiede di rilevare il gene bersaglio dopo un determinato trattamento.Il campione di riferimentoè il campione senza alcun trattamento, che viene spesso definito wild type in biologia.
Repliche sperimentalisono molto importanti.Generalmente, il numero di repliche persuasive deve essere superiore a tre.È necessario distinguere cos'è la replicazione biologica e cos'è la replicazione tecnica.
Repliche biologiche: Lo stesso esperimento di verifica fatto con materiali diversi (tempo, piante, lotti, piastre di reazione).

Duplicazione biologica
Prendiamo come esempio il trattamento antiparassitario del peperone.Vogliamo spruzzare pesticidi sulle tre piante di ABC, quindi le tre piante di ABC sono tre repliche biologiche, e sono lo stesso esperimento di verifica effettuato con materiali diversi.Ma come esperimento, è decisamente necessario un controllo, quindi possiamo spruzzare uno dei rami della pianta A per formare un gruppo sperimentale della pianta A, e non spruzzare gli altri rami della pianta A per formare un gruppo di controllo.Fai lo stesso per B e C.
Repliche tecniche (repliche tecniche): È un esperimento ripetuto progettato per evitare errori causati dall'operazione, che in realtà è un foro duplicato incluso nello stesso materiale.Sia i trattamenti che i controlli devono avere impostazioni di replica (minimo tre) del gene target e del gene di riferimento interno.
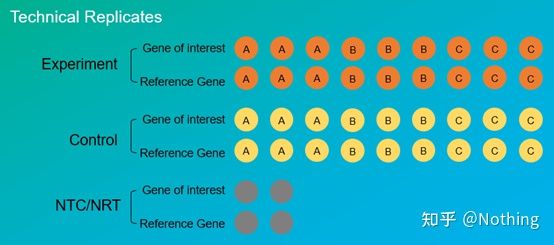
Ripetizione tecnica
Prendiamo di nuovo come esempio il peperone trattato con pesticidi.Per il gruppo sperimentale della pianta A, abbiamo realizzato tre fori PCR di 1, 2 e 3 rispettivamente per il gene bersaglio e il gene di riferimento interno, in modo da ottenere la media dopo il rilevamento.Per il controllo dell'impianto A vengono trattati allo stesso modo anche i Gruppi A.Allo stesso modo, fai lo stesso trattamento per le piante B e C.Questa è ripetizione tecnica.
Vale la pena notare chequello che entra nella statistica è la ripetizione biologica, e la ripetizione tecnica serve a verificare se ci sono fenomeni casuali nel processo sperimentale, in modo da rendere credibili i risultati sperimentali, cioè evitare errori facendone la media come spesso diciamo.
Controlli negativi: NTC e NRT
NTC (controllo senza modello), un controllo senza stampo, viene utilizzato per verificare se il materiale sperimentale è contaminato.Generalmente, l'acqua viene utilizzata come modello.Se c'è una reazione fluorescente, indica che si è verificata una contaminazione da acido nucleico in laboratorio.
Questi inquinamenti provengono da: acqua impura, reagenti non qualificati contenenti DNA endogeno, inquinamento da primer, inquinamento da apparecchiature di laboratorio, inquinamento da aerosol, ecc., è necessario utilizzare scavenger di RNasi e inibitori di RNasi.L'inquinamento da aerosol è il più difficile da trovare.Immagina che il tuo laboratorio sia come lo smog, con vari acidi nucleici sospesi nell'aria.

NRT (senza trascrittasi inversa), il controllo senza trascrizione inversa, è l'RNA trascritto non inverso come controllo negativo, che è il controllo del residuo di gDNA.
Quando si esegue l'espressione genica, la quantità di RNA viene rilevata rilevando la quantità di cDNA dopo la trascrizione inversa.Se c'è un residuo di gDNA quando l'RNA viene purificato, causerà errori nei risultati sperimentali, perché i risultati effettivi ottenuti sono gDNA e cDNA.A livello aggregato, non solo cDNA, il gDNA deve essere completamente rimosso durante l'estrazione dell'RNA.
MIQE (2): informazioni campione
Le cosiddette informazioni sul campione significano che quando pubblichiamo un articolo su qPCR, dobbiamo spiegare chiaramente le informazioni sul campione, che è una parte indispensabile dell'articolo.Allo stesso modo, quando elaboriamo i campioni, dobbiamo anche regolare le nostre operazioni per garantire la validità dei campioni.

La descrizione del campione è solo un risultato, e dovremmo prestare maggiore attenzione ai materiali prelevati durante l'intero esperimento.
Selezione di materiali sperimentali
Campioni di sangue: scegli sangue fresco, non più di 4 ore.Campioni di cellule: scegli di raccogliere cellule fresche in un periodo di crescita vigorosa.Tessuto animale: scegli tessuto fresco e in crescita vigorosa.Tessuto vegetale: scegli tessuti freschi e giovani.
Avrai notato che in queste poche frasi c'è una parola chiave: fresco.
Per i campioni di cui sopra, il kit migliore, economico e stabile sul mercato è il kit di Foregene, che può estrarre rapidamente e facilmente il loro DNA e RNA.
Mini kit per il DNA del sangue
Kit di isolamento dell'RNA totale delle cellule
Kit di isolamento dell'RNA totale animale
Kit di isolamento dell'RNA totale delle piante
Kit di isolamento dell'RNA totale delle piante Plus
Kit di isolamento del DNA vegetale
Stoccaggio di materiali sperimentali
In generale, si sconsiglia di conservare i campioni, se le condizioni lo consentono.Tuttavia, ci sono molti amici che non possono condurre esperimenti subito dopo il campionamento e alcuni hanno persino bisogno di portare sul campo serbatoi di azoto liquido per il campionamento.
Per questo tipo di amico laborioso, posso solo dire che non capisci i materiali di consumo dei reagenti.Ora molte aziende di consumabili di reagenti producono reagenti in grado di conservare campioni di RNA a temperatura ambiente e puoi scegliere di usarli.Il metodo di stoccaggio convenzionale è lo stoccaggio di azoto liquido, utilizzando un piccolo serbatoio di azoto liquido facile da trasportare.Dopo aver riportato il campione in laboratorio, conservarlo in frigorifero a -80°C.

Per gli esperimenti che coinvolgono l'RNA, deve essere seguito il principio delle sei parole:bassa temperatura , senza enzimi ,Eveloce .
Il concetto di bassa temperatura è di facile comprensione;senza enzimi, RNase è ovunque nel mondo in cui viviamo (altrimenti saresti stato ucciso dall'HIV), quindi come evitare RNase quando fai esperimenti è un concetto molto importante;veloce,Non c'è Kung Fu al mondo che non possa essere infranto, solo la velocità non può essere infranta.
Quindi, in un certo senso, più breve è il tempo di estrazione, migliore è il kit.Perché lo faForegeneIl kit di Enfatizza la velocità, perché la conoscono bene.
PS: Alcune ragazze fanno esperimenti con molta attenzione, ma non sono brave come una schiacciata dopo diversi anni di lavoro.Sentono che Dio è ingiusto, si lamentano degli altri e cercano la vita.Infatti lei non lo capiva.Non proteggeva bene l'RNA e il giocatore di schiacciate era agile.Quando stava facendo l'esperimento, pensava che avrebbe finito la schiacciata con tre volte, cinque volte e due divisioni, ma ha fatto bene l'esperimento.
Nota: Più lento, più possibilità di invasione di RNase.Come allenarsi per essere veloci?Non c'è modo, basta esercitarsi di più.
Per esperimenti diversi e campioni diversi, è ancora necessario leggere più letteratura e scegliere un metodo appropriato per l'elaborazione.Per il processo di raccolta e conservazione dei campioni, MIQE richiede che sia chiaramente scritto nel documento, in modo che i revisori possano rivedere l'affidabilità del documento, ed è anche conveniente per i giovani sbalorditi ripetere l'esperimento.
Sebbene gli esperimenti biologici siano difficili, sono di fascia alta.Se non stai attento, puoi ribaltare il mondo.Ad esempio, trasformando la SARS in una crisi biochimica o producendo riso ibrido per salvare 1,3 miliardi di persone.L'immagine qui sotto è un esperimento chimico, dovresti capire quanto sei orgoglioso della tua ricerca solo guardando il suo aspetto simile a un cazzo.Lascia perdere, non infangarlo.

MIQE (3) – estrazione dell'acido nucleico.
L'estrazione dell'acido nucleico è un grande evento e tutti gli esperimenti di biologia molecolare iniziano con l'estrazione dell'acido nucleico.Prima di tutto, copiamo il contenuto del MIQE sull'estrazione dell'acido nucleico.
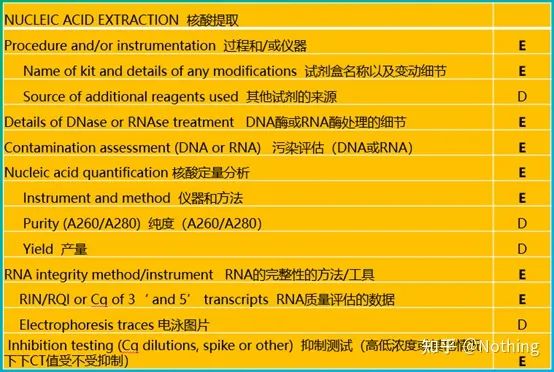
Guardando questo modulo, non puoi rimanere in superficie.La forma è un dogma.Per essere uno studente eccellente, devi chiederti perché.Il contenuto essenziale di questa tabella è: Perseguirela purezza, l'integrità, la consistenza e la quantità di estrazione dell'RNA .
La prima parte delprocesso o strumento è la fase di estrazione dell'acido nucleico.Se si utilizza un estrattore automatico di acido nucleico per estrarre (avanzato, si prega di contattarmi per l'acquisto), è necessario indicare il nome del modello dello strumento.

Il nome del kit e
quale kit è stato utilizzato per i dettagli della modifica, quali reagenti speciali sono stati aggiunti o quali operazioni speciali sono state eseguite dovrebbe essere spiegato chiaramente in modo che altri possano facilmente ripetere l'esperimento.
Alcune persone aggiungono alcuni reagenti speciali durante l'estrazione di campioni speciali, pensando che questa sia la loro arma segreta e non lo dicono agli altri.Pur mantenendolo segreto, perdono anche l'opportunità di far brillare il tuo articolo.Non essere intelligente, devi essere più onesto del vecchio Zhang di campagna nella ricerca scientifica, se vuoi essere intelligente, l'articolo ti renderà stupido.
deve ricordare il numero del prodotto del kitquando ordini il kit e scrivi l'articolo .Generalmente ci sono due numeri sul kit: Cat - numero di catalogo (numero del prodotto, numero dell'articolo), Lot - numero di lotto del prodotto (utilizzato per indicare da quale lotto proviene il prodotto).
Inoltre, il numero CAS viene spesso utilizzato quando si ordinano reagenti biochimici e lo renderò popolare insieme.Il numero CAS è il numero assegnato dall'American Chemical Society a ogni nuovo farmaco chimico.Generalmente, tre numeri sono collegati da un trattino.Numero CAS di Rushui: 7732-18-5.Le sostanze chimiche hanno spesso più pseudonimi, ma il numero CAS è univoco.Quando ordini un medicinale, puoi prima controllarne il numero CAS.
Più vicino a casa, perché dobbiamo descrivere chiaramente queste cose?Infatti serve anche a controllare la qualità dell'estrazione dell'RNA.L'uso di strumenti e kit renderà l'estrazione dell'RNA più coerente.La scala di estrazione dei laboratori ordinari non è grande e può essere ottenuta con i kit.
I dettagli del trattamento DNase o RNase
L'importante questione della PCR quantitativa fluorescente è prevenire la contaminazione del DNA e non sperimentare se c'è contaminazione.Pertanto, è imperativo indicare il processo utilizzato per elaborare il DNA, al fine di dimostrare che il DNA nel processo sperimentale è stato completamente e completamente rimosso.rappresentato da un diagramma schematico.
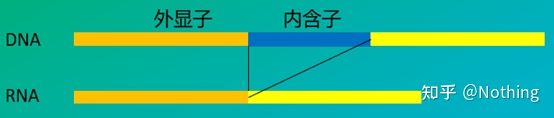
Diagramma schematico di RNA e DNA
In generale, il metodo per rimuovere il DNA consiste nel trattare l'RNA con DNasi dopo l'estrazione.Tuttavia, questi sono metodi relativamente vecchi.I kit commerciali per l'estrazione dell'RNA sono stati in grado di rimuovere il DNA durante il processo di estrazione senza aggiungere DNasi.Ad esempio, una serie di kit di Foregene .
Nota: La rimozione del DNA durante l'estrazione dell'RNA è un'arma a doppio taglio molto pericolosa, che prolungherà il tempo dell'operazione di estrazione dell'RNA e aumenterà il rischio di degradazione dell'RNA.Fondamentalmente, è un compromesso tra resa e purezza dell'RNA.
Inoltre, la quantità di DNasi aggiunta alla colonna di adsorbimento a base di silice è molto ridotta e per ottenere l'effetto è necessario utilizzare DNasi di alta qualità.La DNasi non ottimizzata non può essere digerita rapidamente e completamente.Questo è un test del livello tecnico del commerciante.Naturalmente, ci sono commercianti ancora più strani che si vantano che il DNA può essere rimosso senza DNasi.Si può dire che chiunque si vanti che il DNA può essere completamente rimosso senza DNasi è un teppista.Il DNA è una struttura a doppio filamento relativamente stabile e non può essere spazzato via solo parlando e ridendo.
Valutazione della contaminazione
metodo di valutazione: rilevamento elettroforesi, 1% agarosio, 6 V/cm, 15 min, caricamento 1-3 ul
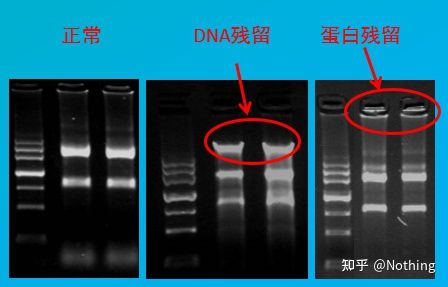
Analisi quantitativa degli acidi nucleici
viene solitamente misurato utilizzando uno spettrofotometro UV.Consentitemi innanzitutto di rendere popolare il significato dei tre valori di OD260, OD280 e OD230.
·OD260nm: è la lunghezza d'onda di assorbimento del massimo picco di assorbimento dell'acido nucleico e il miglior valore misurato varia da 0,1 a 1,0.In caso contrario, diluire o concentrare il campione per portarlo entro il range.
·OD280nm: è la lunghezza d'onda di assorbimento del massimo picco di assorbimento di proteine e sostanze fenoliche.
·OD230nm: è la lunghezza d'onda di assorbimento del massimo picco di assorbimento dei carboidrati.
Successivamente, parliamo del ruolo di ciascun indicatore.Per A260, può essere utilizzato per misurare la resa di acido nucleico.Quando OD260=1, dsDNA=50μg/ml, ssDNA=37μg/ml, RNA=40μg/ml.
Per la purezza, dobbiamo guardare ai rapporti che vediamo comunemente: OD260/280 e OD260/230.
·DNA puro: OD260/280 è approssimativamente uguale a 1,8.Quando è maggiore di 1,9, indica che c'è inquinamento da RNA, e quando è inferiore a 1,6, indica che c'è inquinamento da proteine e fenoli.
·RNA puro: 1.7
·OD260/230: Che si tratti di DNA o RNA, il valore di riferimento è 2,5.Quando è inferiore a 2,0, indica che c'è inquinamento da zucchero, sale e materia organica.
Integrità dell'RNA
È molto importante misurare l'integrità dell'RNA.In generale, è necessario eseguire un esperimento di gel di denaturazione dell'RNA per verificare se la luminosità tra 28S e 18S RNA è una relazione duplice.Quando compare la terza banda 5S, significa che l'RNA ha iniziato a degradarsi, ad eccezione degli invertebrati.

Dati per la valutazione della qualità dell'RNA: oltre ai test di cui sopra, esistono anche alcuni test strumentali più avanzati in termini di integrità dell'RNA, come il test di integrità RQI del sistema di elettroforesi automatica Experion, che può rilevare se l'RNA è degradato in modo invisibile.
Nella ricerca scientifica, la PCR quantitativa fluorescente è un confronto tra il gene bersaglio e il gene di riferimento interno.Pertanto, nel processo di conservazione del campione di RNA, estrazione dell'RNA, ecc., l'obiettivo principale è garantire l'integrità dell'RNA .
Come l'integrità dell'RNA influisce sull'equilibrio tra il gene bersaglio e il gene di riferimento interno può essere facilmente compreso dalla figura sottostante.La degradazione porterà all'incompletezza del gene, che si tratti dell'incompletezza del gene di riferimento interno o dell'incompletezza del gene bersaglio, avrà un grande impatto sui dati.

Il diagramma schematico del gene bersaglio e del gene di riferimento non deve essere vero
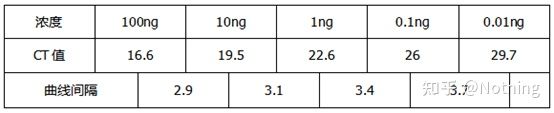
Test di inibizione (se il valore CT viene soppresso ad alta o bassa concentrazione o in altre condizioni)
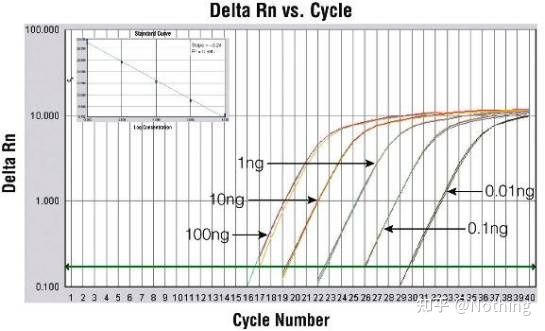
Prendendo questa figura come esempio, i valori Ct delle cinque curve sono i seguenti.La distribuzione dei valori CT tra le curve non è uniforme e i valori Ct sono ritardati in presenza di alte e basse concentrazioni, come nel caso dell'inibizione della PCR.
Punto chiave: nel processo di estrazione dell'RNA, dobbiamo abbandonare le idee sbagliate e stabilire quelle corrette.
L'idea sbagliata è: l'estrazione dell'RNA persegue solo la resa, pensando che maggiore è la quantità di RNA ottenuta, meglio è.Infatti, quando facciamo la quantificazione, se il numero di geni non è molto grande, non abbiamo bisogno di molto RNA.La quantità di RNA che estrai è più che sufficiente.
Il concetto corretto è:L'estrazione dell'RNA dovrebbe perseguire la purezza, l'integrità e la coerenza.La purezza può garantire che la successiva trascrizione inversa non sia inibita e che i dati non vengano influenzati dal DNA.L'integrità garantisce l'equilibrio delle sequenze target e dei riferimenti interni.La coerenza garantisce un caricamento stabile del campione.
MIQE (4) - trascrizione inversa
Idea sbagliata: il perseguimento di un volume di campioni più elevato.
Concetto corretto: Perseguire la coerenza (stabilità), indipendentemente dalla quantità di RNA caricato, l'efficienza della trascrizione inversa rimane coerente, assicurando che le differenze nel cDNA possano riflettere veramente le differenze nell'mRNA.
Spieghiamo questo processo con un diagramma schematico:
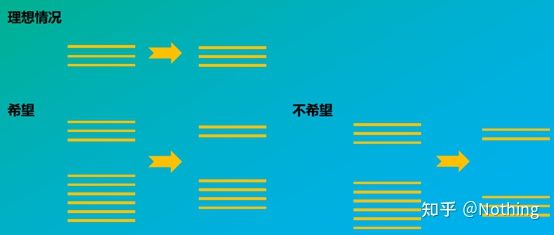
Diagramma schematico dell'efficienza della trascrizione inversa, non essere vero
Prima di tutto, dobbiamo capire la differenza tra il processo di trascrizione inversa e il processo di PCR.La PCR subisce molteplici processi di riscaldamento e ricottura e il frammento target cresce in modo esponenziale;mentre la trascrizione inversa non ha questo processo, possiamo immaginare che la trascrizione inversa sia in realtà uno a uno Durante il processo di replicazione, tanti pezzi di RNA
poiché ci sono molti pezzi di informazioni sul cDNA, dovrebbe essere capito ormai, perché frammenti grandi e piccoli sono stati trascritti al contrario ed è impossibile concentrarsi su un frammento.E poiché la quantità di RNA è relativamente piccola, anche la quantità di cDNA ottenuta è relativamente piccola, a differenza della PCR, che ha un effetto di amplificazione, quindi è praticamente impossibile da rilevare.
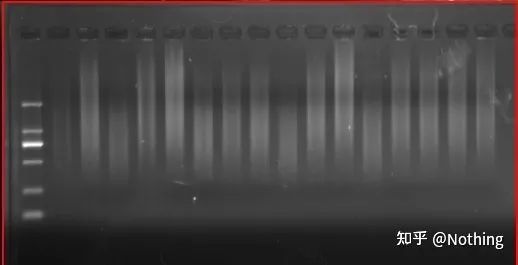
Risultati dell'elettroforesi del cDNA
In secondo luogo, idealmente, la trascrizione inversa viene eseguita uno a uno, ma nessuna trascrittasi inversa di nessuna azienda può ottenere questo effetto.Fondamentalmente, l'efficienza della maggior parte delle trascrittasi inverse oscilla tra il 30 e il 50%.Se questo è il caso, preferiremmo avere un'efficienza di trascrizione inversa relativamente stabile, che è ciò che vogliamo vedere nella figura: 3 RNA ottengono 2 cDNA, 6 RNA ottengono 4 cDNA, quindi non importa quanto campione viene caricato, l'efficienza di trascrizione inversa è relativamente stabile.Non vogliamo vedere la situazione in cui l'efficienza della trascrizione inversa è instabile e l'alta concentrazione è inibita.
Quindi, come verificare se l'efficienza della trascrizione inversa è stabile?Il metodo è molto semplice, devi solo fare un test di confronto: uno è invertire la trascrizione in cDNA dopo aver raddoppiato la diluizione dell'RNA, e l'altro è fare una doppia diluizione dopo aver invertito la trascrizione in cDNA, quindi fare qPCR per vedere la pendenza ottenuta È coerente.Come studente eccellente, dovresti capirlo in pochi secondi.Come mostrato di seguito:
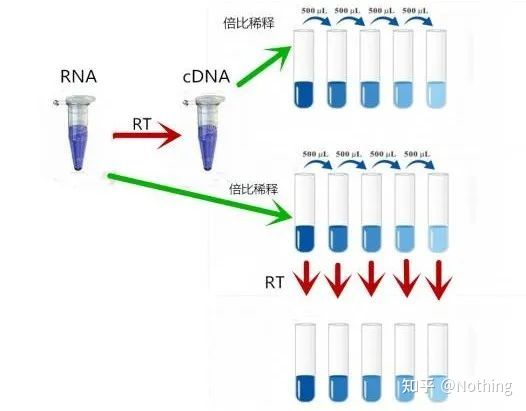
Diluizione di RNA e cDNA per verificare se l'efficienza della trascrizione inversa è stabile
Trascrittasi inversa e kit
In che modo la PCR quantitativa fluorescente perfetta può avere un'eccellente trascrittasi inversa e un kit.La trascrittasi inversa è approssimativamente divisa in due tipi in base alla fonte, AMV oM-MLV, e le loro prestazioni sono le stesse di quelle mostrate nella tabella.
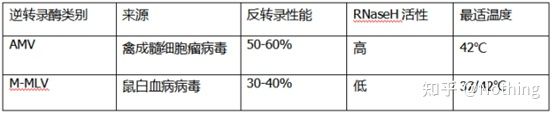
Attività dell'RNasi H
RNasi H è Ribonucleasi H, il nome cinese è ribonucleasi H, che è un'endoribonucleasi che può idrolizzare specificamente l'RNA nella catena ibrida DNA-RNA.L'RNasi H non può idrolizzare i legami fosfodiestere nel DNA o RNA a filamento singolo o doppio, cioè non può digerire DNA o RNA a filamento singolo o doppio.Comunemente utilizzato nella sintesi del secondo filamento di cDNA.
È una cosa strana.Diciamo che la trascrittasi inversa ha attività RNasi H, non che la trascrittasi inversa contiene RNasi H, e potrebbe non essere possibile separare l'RNasi H dalla trascrittasi inversa, forse a causa della conformazione di alcuni gruppi nella trascrittasi inversa Questa attività è causata dalla trascrittasi inversa.
Pertanto, indipendentemente dalla maggiore efficienza di trascrizione inversa di AMV, la sua attività RNasi H riduce la resa di cDNA.Naturalmente, i produttori di reagenti ottimizzano costantemente i loro prodotti per eliminare il più possibile l'attività dell'RNasi H nella trascrittasi inversa per aumentare la resa del cDNA.
Temperatura di ricottura
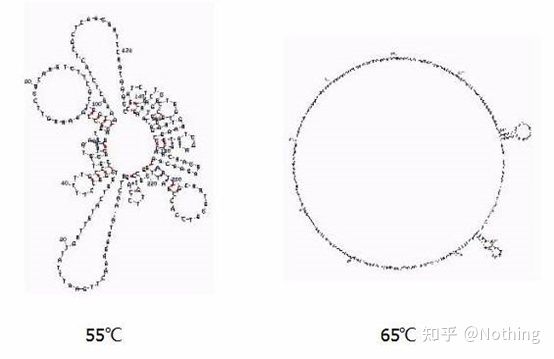
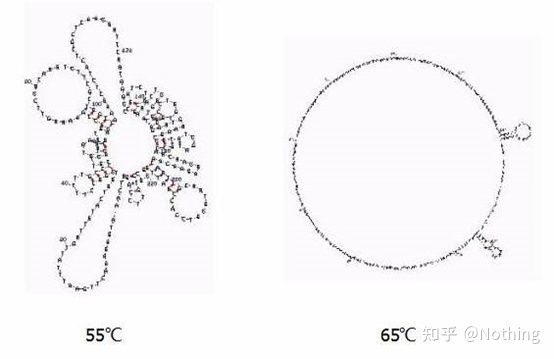
Struttura secondaria dell'RNA a diverse temperature
Vedere la figura sopra per la struttura secondaria dell'RNA a diverse temperature e utilizzare lo strumento online mFold per determinare la struttura secondaria del frammento target in condizioni specifiche di temperatura e concentrazione di sale.A 55°C, la struttura secondaria dell'RNA è ancora molto complessa, la trascrittasi inversa non può funzionare e la struttura secondaria non può essere completamente risolta fino a 65°C, mentre la temperatura ottimale di AMV e M-MLV è molto inferiore a questa temperatura.
cosa fare?La struttura secondaria è l'accoppiamento complementare del modello stesso, che porta a una forte competizione tra il primer e la trascrittasi inversa e il modello, con conseguenti una serie di problemi come basso E e scarsa ripetibilità.
cosa fare?Aumentare la temperatura di ricottura solo il più possibile.
Molti produttori di reagenti stanno migliorando la loro trascrittasi inversa attraverso l'ingegneria genetica.Alcuni aumentano la temperatura di reazione, come Jifan e Aidelai, e alcuni rimuovono il gruppo attivo dell'enzima RNasi H per migliorare l'affinità tra l'enzima e il modello di RNA.L'elevata affinità può spremere in modo competitivo la struttura secondaria e leggerla senza problemi, oltre a migliorare notevolmente l'efficienza della trascrizione inversa.
Punto chiave: la trascrizione inversa è più importante per perseguire la coerenza dell'efficienza della trascrizione inversa (gli enzimi devono essere non solo efficienti ma anche stabili), piuttosto che la quantità di campione caricato, se non è una PCR quantitativa fluorescente su larga scala, non sarà affatto possibile.Più cDNA.
Vari produttori hanno anche compiuto alcuni sforzi nella ricerca della coerenza.Ad esempio, la maggior parte delle aziende ha ora confezionato la trascrizione inversa come kit standard per la vendita, che è una buona scelta.
Ad esempio, i kit RT Easy Series di Foregene:
RT Easy I(Master premix per kit di sintesi cDNA primo filamento)
MIQE (5) – informazioni sul gene bersaglio

La figura sopra spiega
1. Se questo gene è efficace per esperimenti ripetuti può generalmente essere verificato da esperimenti ripetuti.
2. ID genetico, sai.
3. Lunghezza del gene, la lunghezza totale del gene bersaglio non è sicuramente un problema.Quando si progettano i primer, assicurarsi che la lunghezza dell'amplicone sia compresa tra 80 e 200 bp per garantire una migliore efficienza di amplificazione.
4. Informazioni di confronto di sequenza Blast, il gene bersaglio deve essere confrontato nella banca genetica per prevenire l'amplificazione non specifica.
5. Presenza di pseudogeni.Uno pseudogene è una sequenza di DNA simile a un gene normale ma perde la sua normale funzione.Esiste spesso nella famiglia multigenica degli eucarioti.Di solito è rappresentato da ψ.È una copia del DNA genomico non funzionale nel genoma che è molto simile alla sequenza del gene codificante., generalmente non sono trascritti e non hanno un chiaro significato fisiologico.
6. Posizione dei primer rispetto a esoni e introni.Nei primi anni, quando abbiamo risolto il problema della contaminazione del DNA, abbiamo spesso prestato attenzione alle posizioni di primer, esoni e introni e generalmente abbiamo considerato la progettazione di primer attraverso gli introni per evitare l'amplificazione del DNA.Si prega di vedere la figura seguente: il nero rappresenta gli introni, vari blu rappresentano gli esoni, il rosa rappresenta i primer comuni e il rosso vivo rappresenta i primer che si estendono sugli introni.
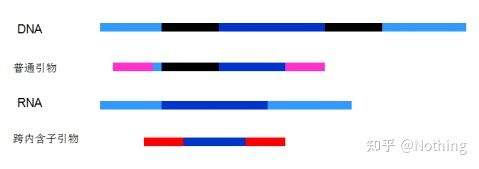
Schematico, mai vero
Sembra un piano perfetto, ma in realtà, nella maggior parte dei casi, i primer trans-introni non sono così magici come immaginato e causeranno anche un'amplificazione non specifica.Quindi il modo migliore per prevenire la contaminazione del DNA è rimuovere completamente il DNA.
7. Previsione della conformazione.Utilizzando di nuovo questo esempio, utilizzare lo strumento online mFold per determinare la struttura secondaria del frammento target a una temperatura e concentrazione di sale specifiche.
Struttura secondaria dell'RNA a diverse temperature
La struttura secondaria è l'accoppiamento complementare del modello stesso, che porterà a una forte concorrenza tra il primer e l'accoppiamento del modello, e le possibilità di legame del primer sono minori, risultando in una serie di problemi come E basso e scarsa ripetibilità.Attraverso la previsione del software, se non ci sono problemi di struttura secondaria, sarebbe fantastico.In tal caso, il nostro articolo di follow-up discuterà in modo specifico su come risolvere questo problema.
MIQE (6)—Oligonucleotidi qPCR

Per la PCR quantitativa fluorescente, la prima cosa con cui lotti ogni giorno è l'estrazione dell'RNA e la seconda potrebbe essere la progettazione del primer.
Prima di tutto, controlliamo ancora le regole sulla progettazione dei primer secondo la lista di controllo MIQE.È così semplice che gli stronzi possono ridere, e possiamo finirlo in una frase: scopri la sequenza e la posizione della sonda primer e il metodo di modifica.Per il metodo di purificazione del primer, la sintesi del primer è attualmente così economica, qPCR è degno di PAGE e metodi di purificazione superiori e le informazioni dello strumento di sintesi non sono importanti.Molte persone fanno primer da decenni e non sanno che il sintetizzatore è ABI3900.
Per quanto riguarda i principi della progettazione dei primer, non è necessario memorizzarli a memoria, perché la maggior parte dei software di progettazione dei primer o degli strumenti online possono occuparsi di questi problemi (strumento online consigliato primer3.ut.ee/) e il 99,999% della progettazione dei primer non viene eseguito manualmente Guarda, l'autore a volte progetta centinaia di primer al giorno, se leggi uno per uno, diventerà strabico.
Basta controllare i seguenti punti dopo che i primer sono stati progettati:
1. Progettare i primer vicino all'estremità 3': nel caso di utilizzo di primer oligo dT per la sintesi del primo filamento di cDNA, considerando l'efficienza della trascrizione inversa e l'integrità dell'RNA, i primer progettati devono essere progettati vicino all'estremità 3' per migliorare l'efficienza dell'amplificazione.Usa un'immagine per spiegare come segue (non c'è modo di capirlo):
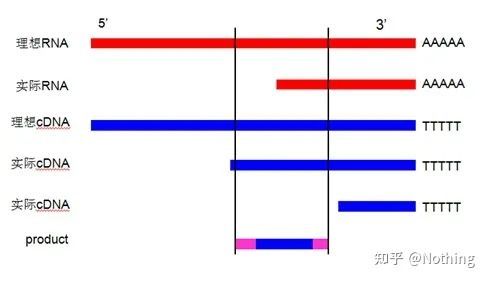
Perché i primer dovrebbero essere progettati vicino all'estremità 3 ′, non deve essere vero
2. Valore TM: il valore Tm è a 55-65°C (poiché l'attività esonucleasica è massima a 60°C) e il contenuto di GC è al 40%-60%.
3. BLAST: per evitare l'amplificazione non specifica del genoma, Blast deve essere utilizzato per la verifica supplementare.
MIQE(7)—processo qPCR
1. Kit qPCR
Secondo i requisiti del MIQE, dobbiamo descrivere chiaramente le condizioni di reazione complete nell'articolo, inclusa la configurazione del sistema di reazione PCR, quale kit viene utilizzato, chi è il produttore, quanto è grande il sistema di reazione, se viene utilizzato il metodo del colorante o il metodo della sonda, le impostazioni del programma PCR.I conducenti veterani scopriranno sicuramente che fintanto che il kit è selezionato, le informazioni di cui sopra sono sostanzialmente determinate.
Allo stato attuale, la fabbricazione e la produzione di kit PCR quantitativi fluorescenti è una tecnologia molto matura.Finché non scegli produttori estremamente scadenti, la probabilità di problemi non è elevata, ma vogliamo comunque condividere con te alcuni punti:
Enzima Taq a caldo:La parte più importante della PCR è l'enzima Taq hot-start.Gli enzimi hot-start sul mercato sono generalmente divisi in due tipi, uno è un enzima hot-start modificato chimicamente (puoi immaginarlo come inclusione in paraffina), e l'altro è un enzima hot-start per la modifica dell'anticorpo (legame antigene-anticorpo).La modificazione chimica è una delle prime modalità di avviamento a caldo degli enzimi.Quando viene raggiunta una certa temperatura, l'enzima rilascerà la sua attività.L'enzima hot-start modificato con anticorpi utilizza metodi biologici per bloccare l'attività dell'enzima.Quando viene raggiunta una certa temperatura, l'anticorpo sarà denaturato e inattivato come proteina, e l'attività enzimatica sarà messa in gioco.
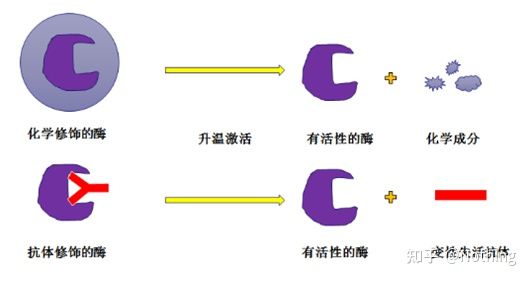
Tuttavia, qual è l'uso di questo?Questo è il caso, l'attività di rilascio degli enzimi modificati con anticorpi è più veloce di quella degli enzimi modificati chimicamente, quindi in termini di sensibilità, gli enzimi modificati con anticorpi hanno un leggero vantaggio, quindi non ci sono praticamente enzimi modificati chimicamente nei kit sul mercato.Se c'è, allora la tecnologia di questo produttore è ancora bloccata nell'era del millennio.
Concentrazione di ioni magnesio:La concentrazione di ioni magnesio è molto importante nella reazione PCR.Un'appropriata concentrazione di ioni magnesio può promuovere il rilascio dell'attività dell'enzima Taq.Se la concentrazione è troppo bassa, l'attività enzimatica sarà significativamente ridotta;se la concentrazione è troppo elevata, l'amplificazione aspecifica catalizzata dall'enzima sarà potenziata.La concentrazione di ioni di magnesio influenzerà anche la ricottura dei primer, la temperatura di fusione del modello e dei prodotti PCR, influenzando così la resa dei frammenti amplificati.La concentrazione di ioni magnesio è generalmente controllata a 25 mM.Naturalmente, per un buon kit, la concentrazione di ioni magnesio deve essere ben controllata.Alcuni commercianti aggiungono un agente chelante di ioni di magnesio al reagente, che può ottenere l'effetto della regolazione automatica della concentrazione di ioni di magnesio.
Concentrazione colorante fluorescente:Il colorante fluorescente, che è il SYBR Green che usiamo di solito, genera principalmente fluorescenza legandosi al solco minore del DNA a doppio filamento, perché il legame del colorante al DNA a doppio filamento non è specifico, vale a dire Finché il DNA a doppio filamento è combinato con esso, può verificarsi fluorescenza, quindi i primer-dimeri e i modelli di DNA nel sistema si combineranno con esso per formare un segnale di fondo.
PS: A causa delle sue proprietà fotosensibili, i prodotti sul mercato sono generalmente confezionati in tubi da centrifuga opachi marroni (come mostrato nella figura sotto).Tuttavia, questo incontrerà un problema.È difficile vedere se il liquido viene aspirato durante il campionamento.A questo proposito, Qingke è davvero il più facile da usare (come mostrato nell'immagine sotto), e il tubo trasparente è confezionato in un sacchetto di latta opaco.Quindi mettilo in un sacchetto di latta, tenendo conto della comodità di evitare la luce e il campionamento.Devi scegliere il numero di prodotto corretto.TSE204 è un'esistenza super conveniente, il che mi fa venire voglia di piantare erba.

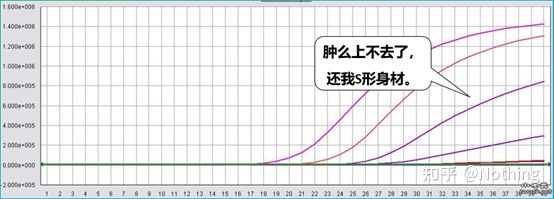
Anche la concentrazione del colorante fluorescente è molto importante.Se la concentrazione è troppo bassa, la curva di amplificazione non salirà nella fase successiva e non sarà perfetta;se la concentrazione è troppo alta, causerà interferenze di rumore.Poiché la PCR quantitativa fluorescente dipende principalmente dal valore CT, se la concentrazione del colorante fluorescente non viene regolata correttamente, il punto più basso è migliore del punto più alto.Naturalmente, la concentrazione di colorante appropriata è la migliore.
ROX: I coloranti ROX vengono utilizzati per correggere gli errori del segnale di fluorescenza da pozzo a pozzo.Alcuni produttori di strumenti richiedono la calibrazione, mentre altri no.Ad esempio, l'uso dello strumento di amplificazione PCR in tempo reale di Thermo Fisher Scientific di solito richiede la calibrazione, inclusi 7300, 7500, 7500Fast, StepOnePlus, ecc. Le istruzioni generali del kit lo descrivono.
Il qPCR Mix di Foregene contiene anche il colorante ROX, utile per l'uso in vari modelli.
Trattamento del legame idrogeno debole: Il trattamento dei legami idrogeno deboli è una questione relativamente tecnica.Nulla ha letto i manuali di molti kit, ma nessuno di loro ha menzionato questo argomento.In effetti, è così importante.La combinazione di basi dipende principalmente dalla forza dei legami idrogeno.I legami idrogeno forti sono un'amplificazione normale e i legami idrogeno deboli portano a un'amplificazione non specifica.Se i legami idrogeno deboli non possono essere eliminati bene, l'amplificazione non specifica non può essere evitata.Nell'ambito dell'autore, solo poche aziende hanno notato questo problema.Quando acquisti il kit, puoi fare riferimento se hai considerato una soluzione in questo senso per il kit che desideri scegliere.
Volume di reazione: Il sistema da 20-50ul è più comunemente usato e volumi più piccoli possono causare errori.In generale, le istruzioni del kit raccomanderanno l'uso di volumi di reazione PCR.Non fare il furbo e usa volumi più piccoli per risparmiare sui costi.l'obiettivo di.Il volume consigliato dai commercianti è stato effettivamente testato e può darsi che non riescano a risolvere il problema degli errori causati da piccoli volumi.
2. Il produttore e il numero di articolo della piastra tubiera
Tutti conoscono il principio della PCR quantitativa fluorescente.La raccolta della fluorescenza viene effettuata principalmente attraverso tappi per provette PCR.Quando si scelgono i consumabili per PCR, prestare attenzione a due punti: buona trasmissione della luce e idoneità allo strumento.In generale vanno bene le schede e le valvole delle marche tradizionali, ma bisogna scegliere con cura in termini di adattamento, altrimenti non si potrà utilizzare lo strumento.

4. Conoscenza di alto livello
MIQE (8)—convalida qPCR
Questa è la massima priorità di qPCR!Così tanti eroi sono caduti nella sabbia qui.Certo, è anche possibile che tu sia fortunato e che i geni che hai studiato siano semplici, quindi hai fluttuato attraverso la grotta di ghiaccio lungo il vento.Le informazioni di verifica di qPCR hanno lo scopo di testare l'affidabilità dei dati.Elenchiamo le informazioni di verifica necessarie come segue:
1.Test di specificità
La specificità dell'amplificazione del gene bersaglio viene testata controllando se l'immagine dell'elettroforesi è una singola banda;verifica del sequenziamento;curva di fusione per vedere se la mappa del picco è singola;verifica della digestione enzimatica e altri metodi.
Qui, ci concentriamo su tL'analisi dell'amplificazione aspecifica con il metodo delle curve di melting.In generale, quando progettiamo i primer, la dimensione del frammento del prodotto deve essere compresa tra 80 e 200 bp, il che rende la temperatura di fusione del prodotto PCR compresa tra 80 e 85 °C.Pertanto, se ci sono picchi vari, devono esserci altri prodotti di amplificazione non specifici;se il picco appare sotto gli 80°C, è generalmente considerato un primer dimero;se il picco appare al di sopra di 85°C, generalmente si considera contaminazione del DNA o altro Amplificazione aspecifica di grandi frammenti.
Nota: a volte c'è un solo picco a 80°C.In questo momento, questo concetto deve essere rispettato.È probabile che i risultati dell'amplificazione siano tutti dimeri di primer.
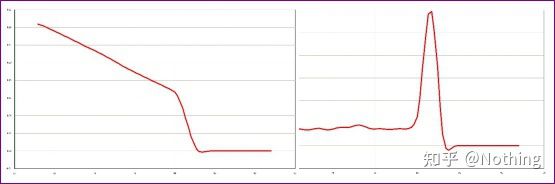
Curva di melting normale (picco singolo senza amplificazione non specifica)
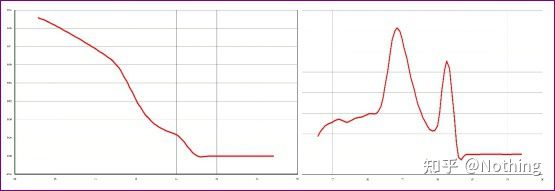
Curva di fusione problematica (amplificazione non specifica di picchi spuri)
【Analisi del caso】
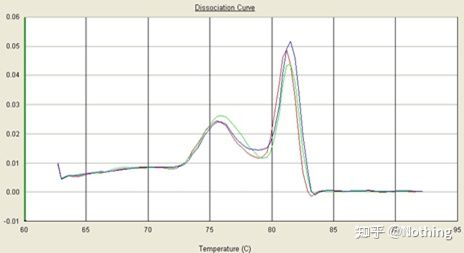
C'è un picco principale, ma il primer dimer è serio
La curva di fusione a picco singolo nella figura sotto può facilmente ingannare i tuoi occhi, pensando che sia un esperimento perfetto, ma il risultato è completamente sbagliato.In questo momento, dobbiamo guardare la temperatura di fusione.La temperatura di picco è inferiore a 80°C, che è completamente primer-dimero.
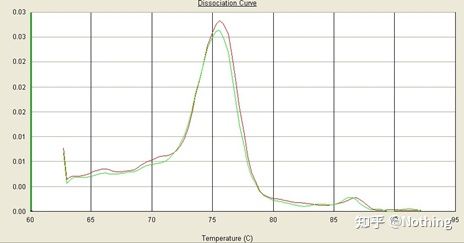
Nessun frammento bersaglio, tutti i dimeri di primer
Ecco, mio fratello non può fermarsi.L'immagine qui sotto è una foto scattata con un cellulare inviatomi da uno stronzo.I reagenti che ha usato sono tutti marchi comunemente usati nel settore.È passato da un marchio con prefisso T a un altro marchio con prefisso T.Penso che tu l'abbia già indovinato.Il sacco di merda mi ha gridato: “Il reagente usato nella prima immagine è troppo buono e il picco è singolo.Successivamente, dopo aver utilizzato il reagente che hai consigliato, diventa come la seconda immagine, con picchi misti.Mi hai reso infelice.“
Separa i due grafici.A prima vista, uno ha un picco singolo e l'altro ha un doppio picco.Sciocchezze, un singolo picco ovviamente va bene.È vero?
Peggio di Dou E, se metto le due immagini nella foto qui sotto, capirai subito.In effetti, siamo facilmente paralizzati da questo tipo di immagine.Dopo un'attenta analisi, abbiamo scoperto che: il picco della prima cifra è a 75°C, che è completamente primer dimer;il picco della seconda figura appare a 75°C e 82°C, almeno ci sono Il prodotto appare.
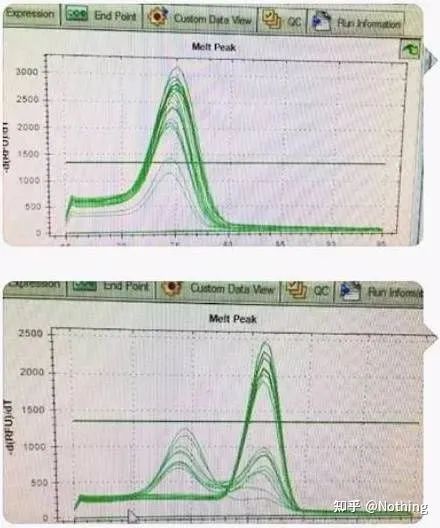
Immagini del feedback degli studenti
Quindi il problema fondamentale non è il problema dei reagenti, ma il problema della progettazione dei primer.Allo stesso tempo, dimostra anche che alcuni grandi marchi non sono di qualità del ferro e dimostra anche ciò che ha detto prima mio fratello: non è il marchio del reagente che supporta il tuo articolo.È il tuo articolo che ha sostenuto la marca di reagenti.Immagina, se il sacco di merda non cambiasse i reagenti, i dati sbagliati verrebbero inviati al diario e ciò che accadrebbe sarebbe una tragedia.
2. Valore Ct del controllo del bianco
Non spiegare, se il controllo del bianco ha un valore Ct, non è inquinamento?Tuttavia, è ancora necessario capire quale controllo del bianco ha un valore Ct.Se è NTC, significa che c'è DNA estraneo come la contaminazione del reagente.Se è NRT, significa che l'RNA estratto ha una contaminazione da DNA.
3. Curva standard
Compresa la pendenza e la formula di calcolo, l'efficienza della PCR può essere calcolata attraverso la formula.Un esperimento perfetto richiede che la pendenza della curva standard si avvicini a 3,32 e che R² si avvicini a 0,9999.
4. Gamma dinamica lineare
Il range dinamico della reazione è lineare.Secondo il modello utilizzato per generare la curva standard, l'intervallo dinamico dovrebbe includere almeno 5 gradienti di concentrazione e prestare attenzione alla variazione dei valori Ct a gradienti di concentrazione elevati e gradienti di concentrazione bassi.
5. Precisione del rilevamento
I cambiamenti nei risultati qPCR, ovvero la scarsa ripetibilità, ovvero la scarsa precisione, sono causati da molti fattori, tra cui temperatura, concentrazione e funzionamento.La precisione qPCR generalmente diventa meno controllabile al diminuire del numero di copie.Idealmente all'interno della variazione sperimentale, questa variazione tecnica dovrebbe essere distinta dalla variazione biologica e le repliche biologiche possono affrontare direttamente le differenze statistiche nei risultati qPCR tra gruppi o trattamenti.In particolare per le analisi diagnostiche, deve essere riportata la migliore precisione inter-analisi (ripetibilità) tra siti e operatori.
6. Efficienza di rilevamento e LOD (in multiplex qPCR)
LOD è la concentrazione più bassa del 95% dei campioni positivi rilevati.In altre parole, la concentrazione di LOD contenuta all'interno di un insieme di replicati del gene bersaglio non dovrebbe superare il 5% delle reazioni fallite.Quando si esegue l'analisi qPCR multiplex, in particolare per il rilevamento simultaneo di mutazioni puntiformi o polimorfismi, la qPCR multiplex deve fornire la prova che l'accuratezza di più frammenti target non è compromessa nella stessa provetta, il rilevamento multiplo e il rilevamento di provetta singola L'efficienza e il LOD dovrebbero essere gli stessi.Soprattutto quando i geni bersaglio ad alta concentrazione e i geni bersaglio a bassa concentrazione vengono simultaneamente amplificati, è necessario prestare attenzione a questo problema.
Problemi e soluzioniIn generale, i problemi spesso riscontrati nel debugging di qPCR si concentrano sui seguenti aspetti:
·amplificazione aspecifica
·Difficile scelta della concentrazione del primer e problemi con i primer-dimeri
·La temperatura di ricottura è imprecisa
·La struttura secondaria influisce sull'efficienza dell'amplificazione
amplificazione non specifica
amplificazione non specificasi verifica , generalmente si considera se il design del primer non è adatto, ma se non hai fretta di cambiare i primer, puoi provare prima i seguenti metodi (il principio è anche allegato):
·Aumentare la temperatura di ricottura – cercare di rendere i legami idrogeno deboli impossibili da mantenere;
· Accorciare i tempi di ricottura e allungamento – ridurre la possibilità di legami idrogeno deboli;
·Ridurre la concentrazione di primer – ridurre la possibilità di legame di primer ridondanti e regioni non target;
Bassa efficienza di amplificazione
La situazione opposta all'amplificazione non specifica - bassa efficienza di amplificazione e le misure per far fronte alla bassa efficienza di amplificazione sono esattamente l'opposto:
· Prolungare il tempo di ricottura e di allungamento;
·Passare alla PCR in tre fasi e ridurre la temperatura di ricottura;
·Aumentare la concentrazione del primer;
Ps: Molti studenti laureati nati negli anni '90 non sono disposti a studiare come eseguire il debug degli esperimenti e sperano che il kit possa risolvere completamente il problema (se vuoi andare in un'azienda di reagenti per fare ricerca e sviluppo dopo la laurea), infatti, anche i produttori di reagenti la pensano in questo modo, spero che sia uno sciocco.Per risolvere facilmente il problema, gli sciocchi devono ancora leggere l'introduzione della società di reagenti per vedere se esiste un fattore che assorbe i legami idrogeno deboli.
Difficile scelta della concentrazione del primer e problemi con i primer-dimeri
Metodo 1: In generale, le istruzioni del kit per qPCR hanno sistemi raccomandati e concentrazioni di primer raccomandate.
Metodo 2: Debug impostando il gradiente di concentrazione del primer.L'immagine qui sotto è stata rubata da un'azienda per illustrare.La figura seguente mostra i risultati quantitativi di fluorescenza ottenuti con tre gradienti di concentrazione di primer (100nM, 250nM, 500nM) e quattro gradienti di concentrazione di template (0,1ng, 1ng, 10ng, 100ng).Il valore Ct dei risultati sperimentali è tracciato come segue:
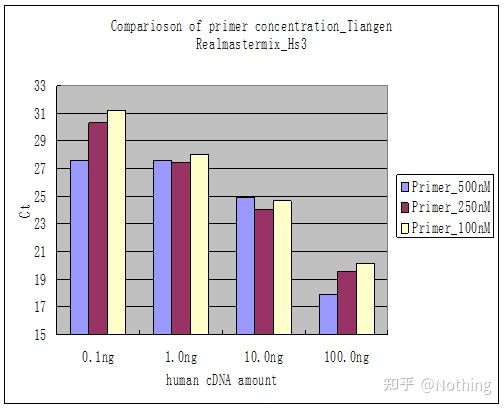
Selezione della concentrazione di primer Concatenare ciascuna concentrazione di primer in una riga come segue:
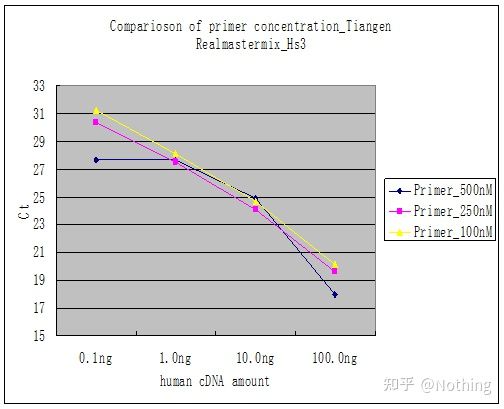
La scelta della concentrazione del primer è ovvia, la relazione lineare della concentrazione del primer di 100nM e 250nM è migliore e la relazione lineare della concentrazione del primer di 500nM è relativamente scarsa.In 100nM e 250nM, il valore Ct di 250nM è relativamente piccolo, quindi la concentrazione ottimale di primer è 250nM.Nella curva di fusione si possono osservare dimeri di primer generalmente gravi.Cosa succede se i primer progettati non possono evitare i primer-dimeri?
Metodo 3: Ridurre la quantità di primer e aumentare la temperatura di ricottura (non è necessario spiegare).
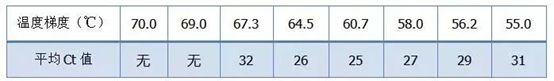
Il valore empirico della temperatura di ricottura è 60°C.Se non sei sicuro, come selezionare una temperatura di ricottura più adatta?La risposta è la stessa della scelta della concentrazione del primer:prova del gradiente.Scatta una foto dalla società Bio-rad per illustrare il problema.Per l'amplificazione di un certo frammento bersaglio, impostare otto gradienti di temperatura, ciascuno con tre ripetizioni, e la curva di amplificazione ottenuta è la seguente:
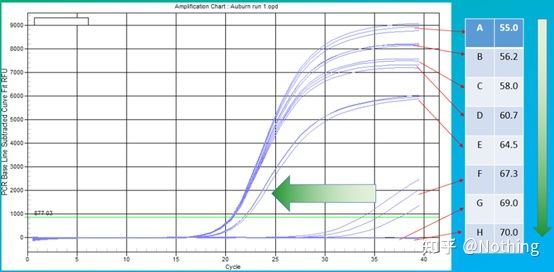
selezione della temperatura di ricottura:
·70°C, 69°C—In pratica, i primer non possono essere combinati, quindi non c'è amplificazione.
·67,3°C – C'è una piccola quantità di amplificazione all'inizio e il valore Ct è relativamente grande.
·64,5°C——Il valore Ct diminuisce.
·A 60,7°C, 58,0°C, 56,2°C e 55,0°C, i valori Ct tendevano sostanzialmente a essere stabili, ma i valori finali di fluorescenza erano diversi.
Come scegliere?Principio: il primo principio è il valore Ct più alto.Per lo stesso valore Ct, scegliere una temperatura di ricottura più alta per evitare la dimerizzazione e l'amplificazione non specifica.Sebbene vi sia un valore di fluorescenza più elevato a 55°C, potrebbero esserci dei dimeri o un'amplificazione non specifica.
Ma se sei intelligente come te, penserai sicuramente: logicamente parlando, se la reazione PCR è molto specifica, fintanto che la concentrazione del primer supera il requisito minimo, i punti alto e basso non dovrebbero avere alcun effetto, proprio come i coloranti fluorescenti e i dNTP.Infatti, fintanto che la temperatura di ricottura è ottimizzata correttamente, l'effetto della concentrazione del primer sul valore Ct sarà naturalmente ridotto al minimo.
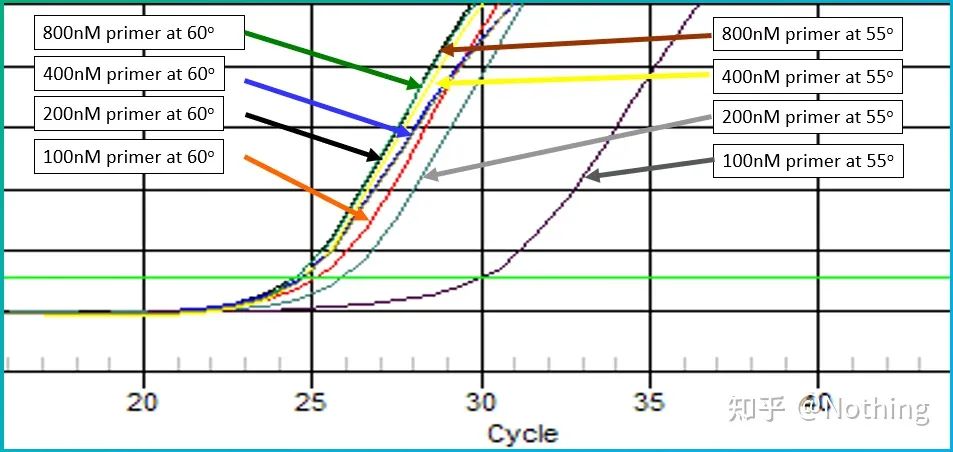
La temperatura di ricottura è ottimizzata correttamente e l'effetto della concentrazione del primer su CT sarà ridotto al minimo
La struttura secondaria influisce sull'efficienza dell'amplificazione
Prendiamo la foto da Bio-rad per illustrare il problema.Progetta anche un gradiente di temperatura per amplificare un gene con una struttura secondaria.
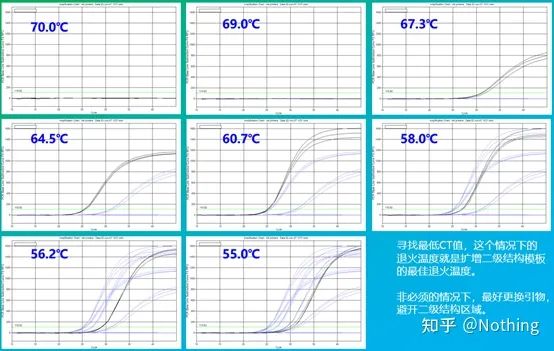
Emerge la struttura secondaria
Si può vedere che al diminuire del gradiente di temperatura iniziano ad apparire i prodotti e il valore Ct avanza, raggiungendo il valore minimo a 60,7°C, e poi al diminuire del gradiente di temperatura, il valore Ct aumenta.Al contrario, all'aumentare della temperatura, la struttura secondaria si apre e l'efficienza di amplificazione aumenta.Dopo aver raggiunto una certa temperatura, l'aumento della temperatura non può migliorare l'efficienza dell'amplificazione.Perché i primer non possono essere combinati stabilmente in questo momento.Perciò,cercare la temperatura con il valore Ct più basso, che è la temperatura migliore per amplificare il modello della struttura secondaria!Naturalmente, gli sciocchi intelligenti devono sapere che se non è necessario, è meglio cambiare i primer ed evitare la regione della struttura secondaria.
5. Livello di applicazione
MIQE—Analisi dei dati

L'analisi dei dati è fornita principalmente dallo strumento PCR quantitativo fluorescente.Nell'articolo precedente è stato svolto molto lavoro di analisi dei dati, come il controllo del bianco, che è stato spiegato nella progettazione dell'esperimento.I geni di riferimento interni, i numeri di ripetizione, ecc. sono stati chiariti., qui spieghiamo principalmente l'applicazione di qPCR.
qPCR è ampiamente utilizzato e la verifica sperimentale e la diagnosi dell'acido nucleico sono gli scenari più comunemente utilizzati.
quantificazione assoluta
Log (concentrazione iniziale) ha una relazione lineare con il numero di cicli.È possibile tracciare una curva standard da uno standard con numero di copie iniziale noto, ovvero è possibile ottenere la relazione lineare della reazione di amplificazione.In base al valore Ct del campione, è possibile calcolare la concentrazione nel campione.La quantità di modelli da includere.
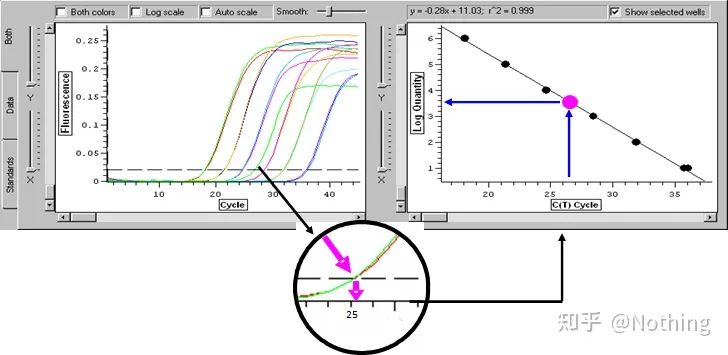
Metodo di calcolo quantitativo assoluto
La quantificazione assoluta deve essere basata sulla curva standard.Per creare una curva standard, è necessario uno standard.Di solito, lo standard è un plasmide ottenuto clonando il gene bersaglio.Perché è un plasmide?Perché il DNA plasmidico circolare è il più stabile.Diluire il prodotto standard in 5-6 gradienti secondo il rapporto di raddoppio (diluizione 10 volte) e prestare attenzione all'uniformità durante la diluizione.Lascia che il valore Ct scenda tra 15 e 30.
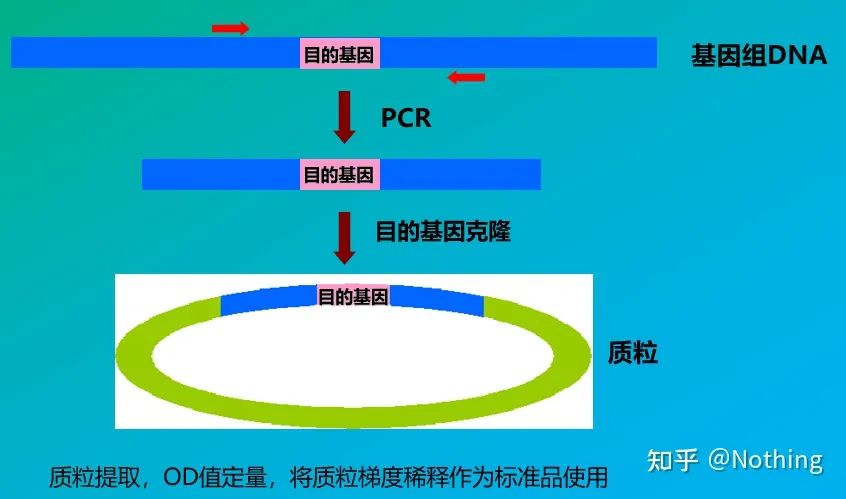
Preparazione standard
Allo stesso tempo, anche il campione da testare dovrebbe essere diluito di conseguenza (ricordare il fattore di diluizione) e anche il valore Ct dovrebbe essere compreso tra 15 e 30.Il prodotto standard + il campione da testare vengono messi insieme sulla macchina.Dopo la corsa, è stata realizzata una curva standard con la sostanza standard ei campioni da testare sono stati portati nella curva standard per calcolare la concentrazione.
La quantificazione dell'HBV del virus dell'epatite B è una tipica quantificazione assoluta, che può calcolare il numero di copie del virus in 1 ml di sangue.
Calcolo del numero di copie
Concentrazione del campione da testare (ng/ul) = OD260 × 50 ug/ml × fattore di diluizione
Peso molecolare del campione = numero di basi × 324
Il numero di copie del campione da testare (copie/ul) = la concentrazione del campione da testare / il peso molecolare del campione × 6 × 1014

Metodo di calcolo del numero di copie
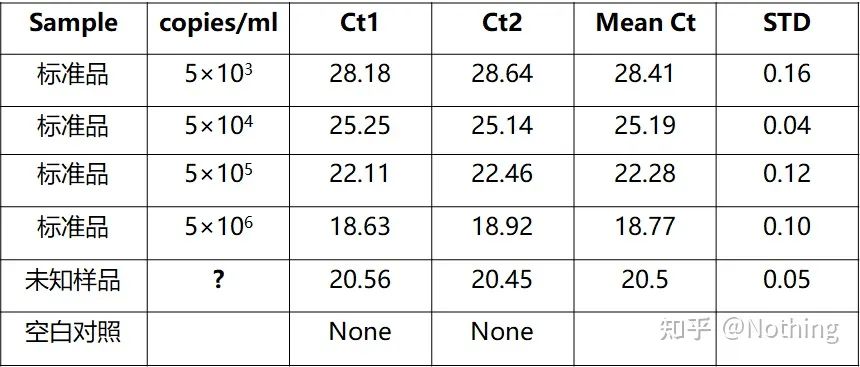

Quanto sopra è il metodo di calcolo per determinare la quantità.Questo è un problema matematico che può essere risolto dopo il diploma di scuola media e i problemi matematici sono generalmente risolti dai computer.Se non capisci, puoi venire a comunicare.
relativa quantificazione
La quantificazione relativa è utilizzata principalmente nella ricerca scientifica.Quanti virus ci sono in 1 ml di sangue, ed è un virus a DNA, questo è un evento relativamente deterministico: la quantità di sangue può essere determinata e il virus a DNA è relativamente stabile.Tuttavia, è difficile per noi confrontare il numero di copie di trascrizione di un determinato gene in una foglia, perché è difficile determinare le dimensioni, il peso e la tenerezza della foglia, la quantità di RNA estratto è difficile da determinare e anche l'efficienza della trascrizione inversa è difficile da determinare, vale a dire, qualsiasi passaggio può rendere i dati sperimentali con errori e non possono essere utilizzati.
Pertanto, la quantificazione relativa deve introdurre un elemento:il gene di riferimento interno.
In altre parole, la quantificazione relativa è in realtà un confronto tra il gene bersaglio e il gene di riferimento interno.Rispetto allo stesso tessuto e alla stessa cellula, l'influenza della dimensione del campione, della quantità di estrazione dell'RNA, dell'efficienza della trascrizione inversa e dell'efficienza della PCR è relativamente piccola.A causa delle ridotte dimensioni del campione, sia i geni di riferimento interni che i geni bersaglio erano relativamente ridotti.Questo è il motivo per cui in precedenza abbiamo enfatizzato l'uniformità e la stabilità.
I geni di riferimento interni sono generalmentegeni domestici( geni domestici ), che si riferiscono a una classe di geni espressi stabilmente in tutte le cellule e i loro prodotti sono necessari per mantenere le attività vitali di base delle cellule.
Non confondere questo concetto.I geni domestici sono termini di funzioni biologiche, mentre i geni di riferimento interni sono termini tecnici sperimentali.I geni domestici devono superare la convalida prima di poter essere selezionati come geni di riferimento interni.
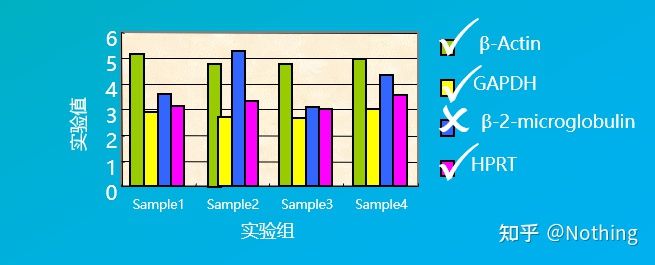
Ad esempio, abbiamo selezionato diversi geni domestici nella figura sottostante per testare i loro livelli di espressione in diverse cellule tissutali e abbiamo scoperto che i livelli di espressione della β-2-microglobulina erano molto diversi da quelli degli altri tre geni, quindi non potevano essere usati come geni di riferimento interni.
Dopo aver compreso la funzione di correzione del gene di riferimento interno, vengono derivati due algoritmi a causa dell'introduzione del gene di riferimento interno.
·metodo della doppia curva standard
·2 – △△metodo Ct (metodo di confronto del valore CT)
Se sei interessato allo studio delle specie e delle funzioni dei geni, per favore abbandona la ricerca sugli algoritmi e usa direttamente le formule, o usa direttamente le macchine;se sei un ragazzo etero in matematica e ingegneria, sentiti libero.
metodo della doppia curva standard
Quantificare il gene target e il gene housekeeping del campione di controllo e del campione da testare attraverso la curva standard, quindi calcolare il valore relativo in base alla formula di calcolo, che è il livello di espressione relativo.
Vantaggi: analisi semplice, ottimizzazione sperimentale relativamente semplice
Svantaggio: per ogni gene, ogni ciclo di esperimenti deve creare una curva standard
Applicazione: uno dei due metodi quantitativi relativi più comunemente usati e riconosciuti nello studio della regolazione dell'espressione genica
La formula è la seguente:

Gli esempi sono i seguenti:
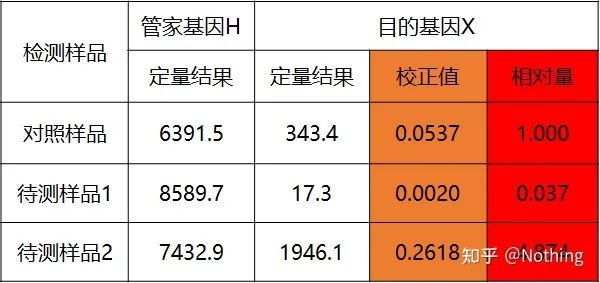
Calcolare l'importo relativo in base al risultato quantitativo
2 – Metodo △△Ct (metodo di confronto del valore CT)

Vantaggi: non è necessario creare una curva standard
Svantaggi: si presume che l'efficienza di amplificazione sia vicina al 100%;la deviazione standard è < 5% e si presume che la curva standard e l'efficienza tra ciascuna amplificazione siano coerenti;l'ottimizzazione delle condizioni sperimentali è più complicata.
Applicazione: uno dei due metodi quantitativi relativi più comunemente usati e riconosciuti nello studio della regolazione dell'espressione genica
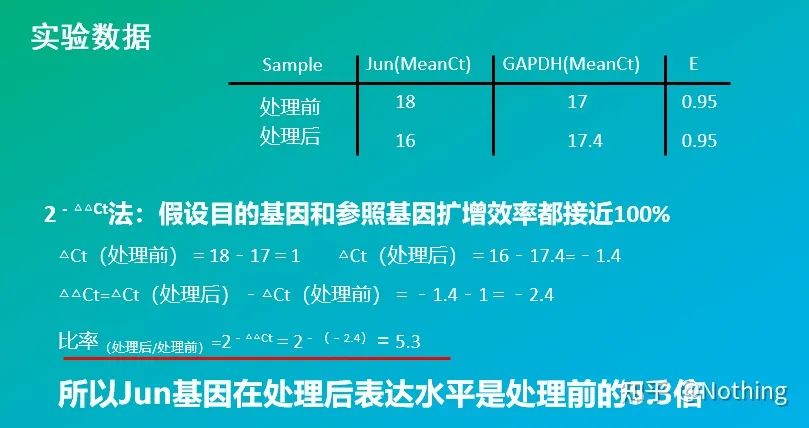
Naturalmente, l'efficienza di amplificazione di solito è impossibile da essere perfettamente 1. Metodo di correzione: se sappiamo che il gene target e il gene di riferimento hanno la stessa efficienza di amplificazione, ma l'efficienza di amplificazione non è uguale a 1, allora 2-△△Ct può essere corretto come: (1+E )-△△Ct, ad esempio, se l'efficienza di amplificazione è 0,95, allora la formula di calcolo può essere corretta a 1,95- △△Ct
Finora, il contenuto sulla PCR quantitativa fluorescente è giunto al termine.
Tempo di pubblicazione: aprile-06-2023



